

Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans
- PMID: 20980553
- PMCID: PMC3106331
- DOI: 10.1101/gr.114777.110
Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans
Abstract
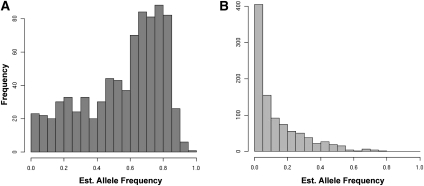
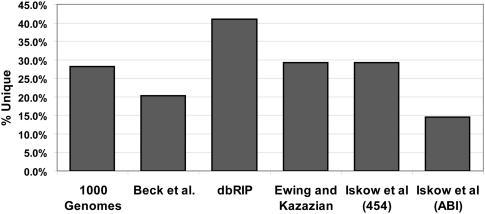
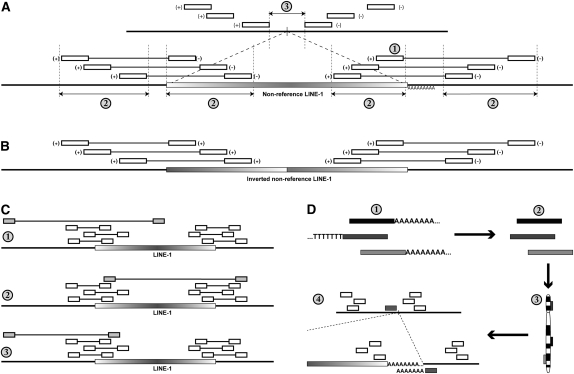
High-throughput sequencing has recently begun to revolutionize the study of structural variants in the genomes of humans and other species. More recently, this technology and others have been applied to the study of human retrotransposon insertion polymorphisms (RIPs), yielding an unprecedented catalog of common and rare variants due to insertional mutagenesis. At the same time, the 1000 Genomes Project has released an enormous amount of whole-genome sequence data. In this article, we present evidence for 1016 L1 insertions across all studies to date that are not represented in the reference human genome assembly, many of which appear to be specific to populations or groups of populations, particularly Africans. Additionally, a cross-comparison of several studies shows that, on average, 27% of surveyed nonreference insertions is present in only one study, indicating the low frequency of many RIPs.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources