

Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads
- PMID: 20980556
- PMCID: PMC3106326
- DOI: 10.1101/gr.111120.110
Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads
Abstract
High-volume sequencing of DNA and RNA is now within reach of any research laboratory and is quickly becoming established as a key research tool. In many workflows, each of the short sequences ("reads") resulting from a sequencing run are first "mapped" (aligned) to a reference sequence to infer the read from which the genomic location derived, a challenging task because of the high data volumes and often large genomes. Existing read mapping software excel in either speed (e.g., BWA, Bowtie, ELAND) or sensitivity (e.g., Novoalign), but not in both. In addition, performance often deteriorates in the presence of sequence variation, particularly so for short insertions and deletions (indels). Here, we present a read mapper, Stampy, which uses a hybrid mapping algorithm and a detailed statistical model to achieve both speed and sensitivity, particularly when reads include sequence variation. This results in a higher useable sequence yield and improved accuracy compared to that of existing software.
Figures
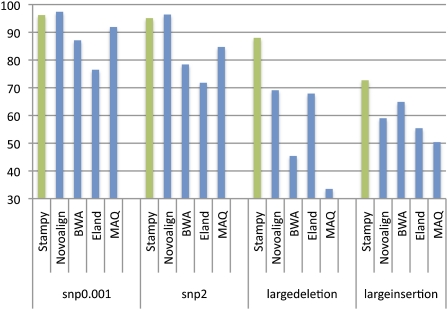
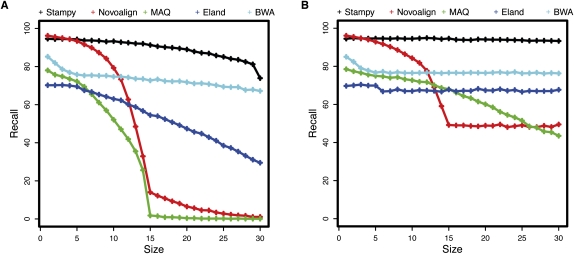
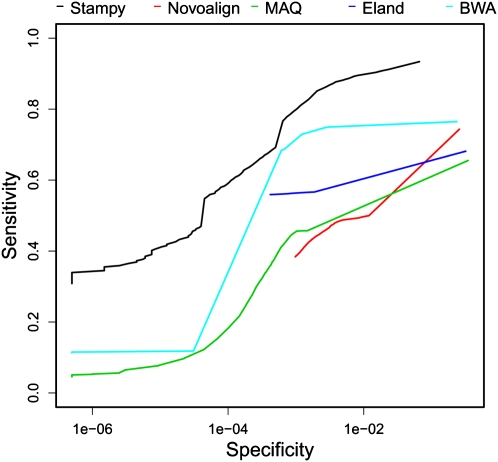
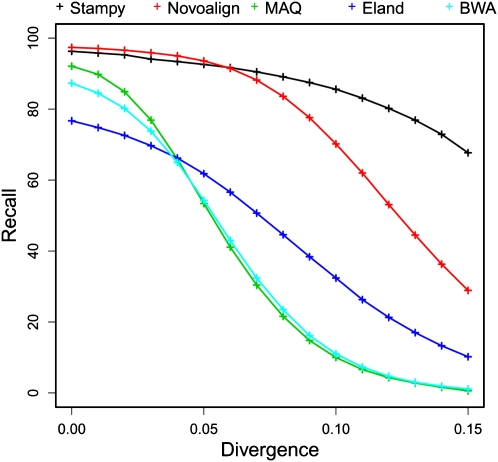
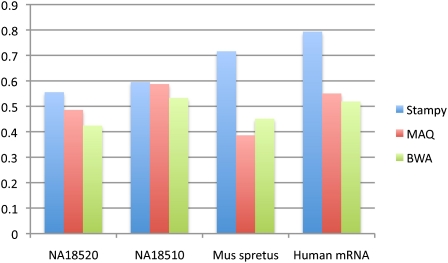
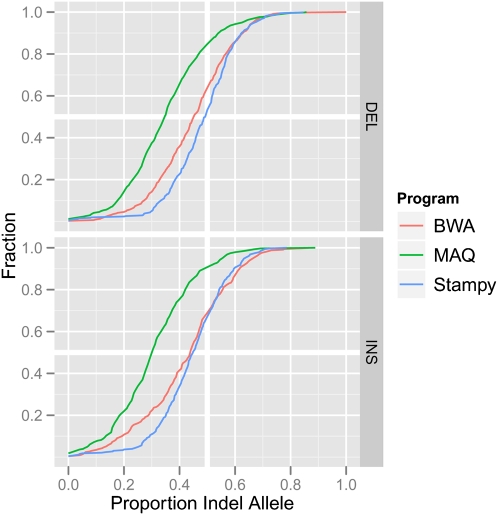
References
-
- Cox AJ 2007. ELAND: Efficient large-scale alignment of nucleotide databases. Illumina, San Diego
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases