

Regulation of benzo[a]pyrene-mediated DNA- and glutathione-adduct formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in human lung cells
- PMID: 21028851
- PMCID: PMC3021323
- DOI: 10.1021/tx100297z
Regulation of benzo[a]pyrene-mediated DNA- and glutathione-adduct formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in human lung cells
Abstract
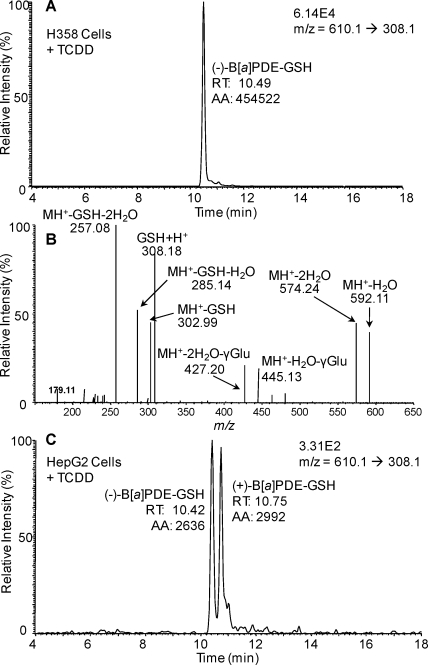
Environmental carcinogens, such as polycyclic aromatic hydrocarbons (PAHs), require metabolic activation to DNA-reactive metabolites in order to exert their tumorigenic effects. Benzo[a]pyrene (B[a]P), a prototypic PAH, is metabolized by cytochrome P450 (P450) 1A1/1B1 and epoxide hydrolase to (-)-B[a]P-7,8-dihydro-7,8-diol (B[a]P-7,8-dihydrodiol). B[a]P-7,8-dihydrodiol then undergoes further P4501A1/1B1-mediated metabolism to the ultimate carcinogen, (+)-anti-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydro-B[a]P (B[a]PDE), which forms DNA-adducts primarily with 2'-deoxyguanosine (dGuo) to form (+)-anti-trans-B[a]PDE-N(2)-dGuo (B[a]PDE-dGuo) in DNA. Pretreatment of cells with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is known to induce P4501A1/1B1 mRNA expression through the aryl hydrocarbon receptor (AhR) pathway. This causes increased B[a]PDE-dGuo formation in liver cells. In contrast, TCDD induction of H358 lung cells surprisingly caused a decrease in (-)-B[a]P-7,8-dihydrodiol-mediated (+)-B[a]PDE-dGuo adduct formation when compared with the non-TCDD-induced cells. Furthermore, treatment of the TCDD-induced cells with (±)-B[a]PDE also resulted in decreased (+)-B[a]PDE-dGuo adduct formation when compared with the non-TCDD-induced cells. These data suggested that it was a detoxification pathway that had been up-regulated rather than an activation pathway that had been down-regulated. LC-MS was used to analyze B[a]PDE-dGuo and B[a]PDE-GSH-adducts in H358 lung and HepG2 liver cells. There was a significant increase in the (-)-B[a]PDE-GSH-adduct with high enantiomeric excess after treatment of the TCDD-induced H358 cells with (±)-B[a]PDE when compared with the noninduced cells. This could explain why increased activation of (-)-B[a]P-7,8-dihydrodiol through TCDD up-regulation of P4501A1/1B1 did not lead to increased (+)-B[a]PDE-dGuo adducts in the H358 lung cells. In addition, TCDD did not induce B[a]PDE-GSH-adduct formation in HepG2 liver cells. (±)-B[a]PDE-GSH-adducts were formed at much lower levels in both TCDD-induced and noninduced HepG2 cells when compared with (-)-B[a]PDE-GSH-adducts in the H358 lung cells. Therefore, our study has revealed that there is a subtle balance between activation and detoxification of B[a]P in lung-derived compared with liver-derived cells and that this determines how much DNA damage occurs.
Figures










Similar articles
-
Aldo-keto reductase- and cytochrome P450-dependent formation of benzo[a]pyrene-derived DNA adducts in human bronchoalveolar cells.Chem Res Toxicol. 2007 Mar;20(3):424-31. doi: 10.1021/tx060180b. Epub 2007 Feb 13. Chem Res Toxicol. 2007. PMID: 17295519
-
Multidrug resistance protein (MRP) 4 attenuates benzo[a]pyrene-mediated DNA-adduct formation in human bronchoalveolar H358 cells.Toxicol Lett. 2012 Feb 25;209(1):58-66. doi: 10.1016/j.toxlet.2011.11.021. Epub 2011 Nov 30. Toxicol Lett. 2012. PMID: 22155354 Free PMC article.
-
Competing roles of cytochrome P450 1A1/1B1 and aldo-keto reductase 1A1 in the metabolic activation of (+/-)-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene in human bronchoalveolar cell extracts.Chem Res Toxicol. 2005 Feb;18(2):365-74. doi: 10.1021/tx0497245. Chem Res Toxicol. 2005. PMID: 15720144
-
Benzo[a]pyrene-Environmental Occurrence, Human Exposure, and Mechanisms of Toxicity.Int J Mol Sci. 2022 Jun 6;23(11):6348. doi: 10.3390/ijms23116348. Int J Mol Sci. 2022. PMID: 35683027 Free PMC article. Review.
-
Influences of polycyclic aromatic hydrocarbon on the epigenome toxicity and its applicability in human health risk assessment.Environ Res. 2022 Oct;213:113677. doi: 10.1016/j.envres.2022.113677. Epub 2022 Jun 14. Environ Res. 2022. PMID: 35714684 Review.
Cited by
-
New insights of CYP1A in endogenous metabolism: a focus on single nucleotide polymorphisms and diseases.Acta Pharm Sin B. 2020 Jan;10(1):91-104. doi: 10.1016/j.apsb.2019.11.016. Epub 2019 Dec 2. Acta Pharm Sin B. 2020. PMID: 31998606 Free PMC article. Review.
-
Polymorphisms in DNA repair genes of XRCC1, XPA, XPC, XPD and associations with lung cancer risk in Chinese people.Thorac Cancer. 2014 May;5(3):232-42. doi: 10.1111/1759-7714.12073. Epub 2014 Apr 22. Thorac Cancer. 2014. PMID: 26767006 Free PMC article.
-
Cigarette smoking enhances the metabolic activation of the polycyclic aromatic hydrocarbon phenanthrene in humans.Carcinogenesis. 2021 Apr 30;42(4):570-577. doi: 10.1093/carcin/bgaa137. Carcinogenesis. 2021. PMID: 33319219 Free PMC article.
-
Long-Term Storage Study of the Certified 1R6F Reference Cigarette.Chem Res Toxicol. 2023 Apr 17;36(4):685-690. doi: 10.1021/acs.chemrestox.3c00004. Epub 2023 Mar 16. Chem Res Toxicol. 2023. PMID: 36926865 Free PMC article.
-
Human Family 1-4 cytochrome P450 enzymes involved in the metabolic activation of xenobiotic and physiological chemicals: an update.Arch Toxicol. 2021 Feb;95(2):395-472. doi: 10.1007/s00204-020-02971-4. Epub 2021 Jan 18. Arch Toxicol. 2021. PMID: 33459808 Free PMC article. Review.
References
-
- Baird W. M.; Hooven L. A.; Mahadevan B. (2005) Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ. Mol. Mutagen. 45, 2−3106–114. - PubMed
-
- Hecht S. S. (2006) Cigarette smoking: cancer risks, carcinogens, and mechanisms. Langenbecks Arch. Surg. 391, 6603–613. - PubMed
-
- Ruan Q.; Gelhaus S. L.; Penning T. M.; Harvey R. G.; Blair I. A. (2007) Aldo-keto reductase- and cytochrome P450-dependent formation of benzo[a]pyrene-derived DNA-adducts in human bronchoalveolar cells. Chem. Res. Toxicol. 20, 3424–431. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
