

The role of autophagy in β-cell lipotoxicity and type 2 diabetes
- PMID: 21029295
- PMCID: PMC3786363
- DOI: 10.1111/j.1463-1326.2010.01268.x
The role of autophagy in β-cell lipotoxicity and type 2 diabetes
Abstract
Autophagy, a ubiquitous catabolic pathway involved in both cell survival and cell death, has been implicated in many age-associated diseases. Recent findings have shown autophagy to be crucial for proper insulin secretion and β-cell viability. Transgenic mice lacking autophagy in their β-cells showed decreased β-cell mass and suppressed glucose-stimulated insulin secretion. Several studies showed that stress can stimulate autophagy in β-cells: the number of autophagosomes is increased in different in vivo models for diabetes, such as db/db mice, mice fed high-fat diet, pdx-1 knockout mice, as well as in in vitro models of glucotoxicity and lipotoxicity. Pharmacological and molecular inhibition of autophagy increases the susceptibility to cell stress, suggesting that autophagy protects against diabetes-relevant stresses. Recent findings, however, question these conclusions. Pancreases of diabetics and β-cells exposed to fatty acids show accumulation of abnormal autophagosome morphology and suppression of lysosomal gene expression suggesting impairment in autophagic turnover. In this review we attempt to give an overview of the data generated by others and by us in view of the possible role of autophagy in diabetes, a role which depending on the conditions, could be beneficial or detrimental in coping with stress.
© 2010 Blackwell Publishing Ltd.
Conflict of interest statement
The authors do not declare any conflict of interest relevant to this manuscript.
Figures
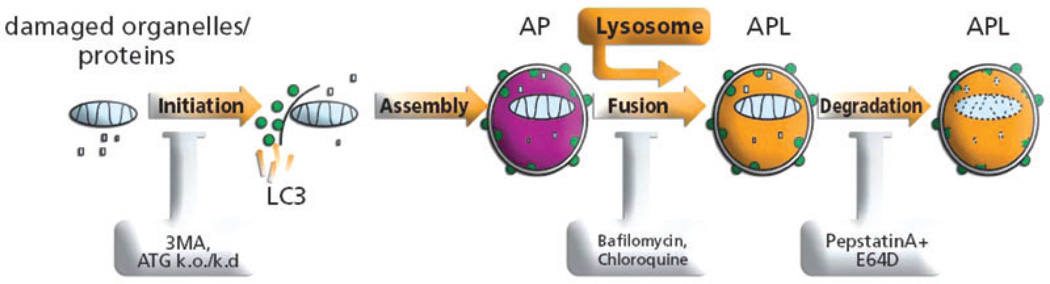
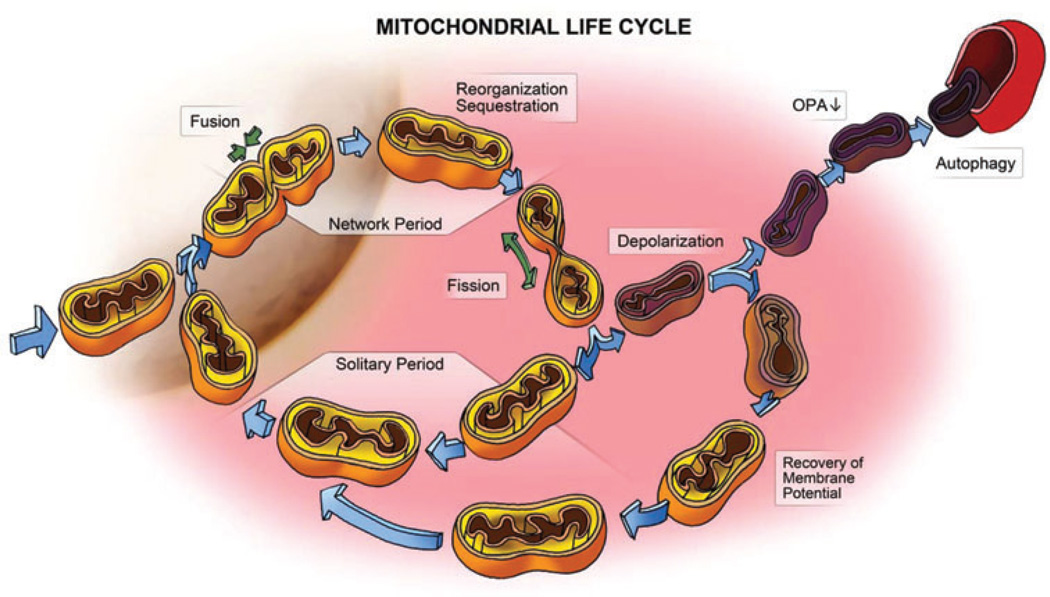
References
-
- Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. - PubMed
-
- Sako Y, Grill VE. A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology. 1990;127:1580–1589. - PubMed
-
- Zhou YP, Grill V. Long term exposure to fatty acids and ketones inhibits B-cell functions in human pancreatic islets of Langerhans. J Clin Endocrinol Metab. 1995;80:1584–1590. - PubMed
-
- Elks ML. Chronic perifusion of rat islets with palmitate suppresses glucose-stimulated insulin release. Endocrinology. 1993;133:208–214. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
