

The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes
- PMID: 21029779
- PMCID: PMC3043253
- DOI: 10.1016/j.jsb.2010.10.014
The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes
Abstract
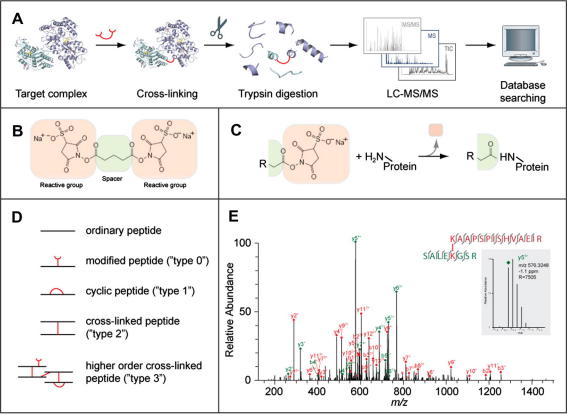
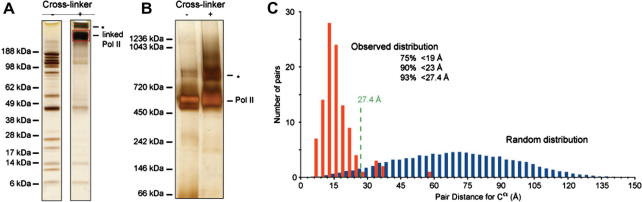
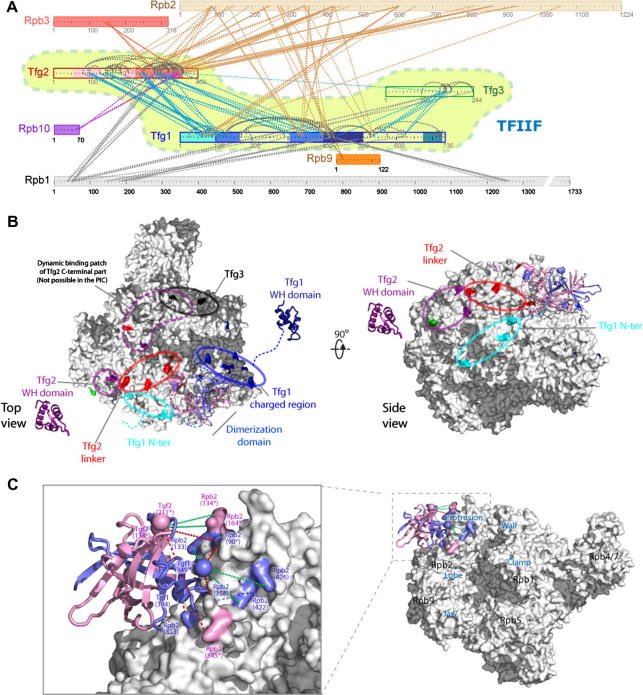
After more than a decade of method development, cross-linking in combination with mass spectrometry and bioinformatics is finally coming of age. This technology now provides improved opportunities for modelling by mapping structural details of functional complexes in solution. The structure of proteins or protein complexes is ascertained by identifying amino acid pairs that are positioned in close proximity to each other. The validity of this technique has recently been benchmarked for large multi-protein complexes, by comparing cross-link data with that from a crystal structure of RNA polymerase II. Here, the specific nature of this cross-linking data will be discussed to assess the technical challenges and opportunities for model building. We believe that once remaining technological challenges of cross-linking/mass spectrometry have been addressed and cross-linking/mass spectrometry data has been incorporated into modelling algorithms it will quickly become an indispensable companion of protein and protein complex modelling and a corner-stone of integrated structural biology.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Alber F., Dokudovskaya S., Veenhoff L.M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B.T., Rout M.P., Sali A. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. - PubMed
-
- Alber F., Dokudovskaya S., Veenhoff L.M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B.T., Sali A., Rout M.P. The molecular architecture of the nuclear pore complex. Nature. 2007;450:695–701. - PubMed
-
- Armache K.J., Mitterweger S., Meinhart A., Cramer P. Structures of complete RNA polymerase II and its subcomplex, Rpb4/7. J. Biol. Chem. 2005;280:7131–7134. - PubMed
-
- Back J.W., de Jong L., Muijsers A.O., de Koster C.G. Chemical cross-linking and mass spectrometry for protein structural modeling. J. Mol. Biol. 2003;331:303–313. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
