

Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A)
- PMID: 21030442
- PMCID: PMC3045621
- DOI: 10.1093/nar/gkq994
Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A)
Abstract
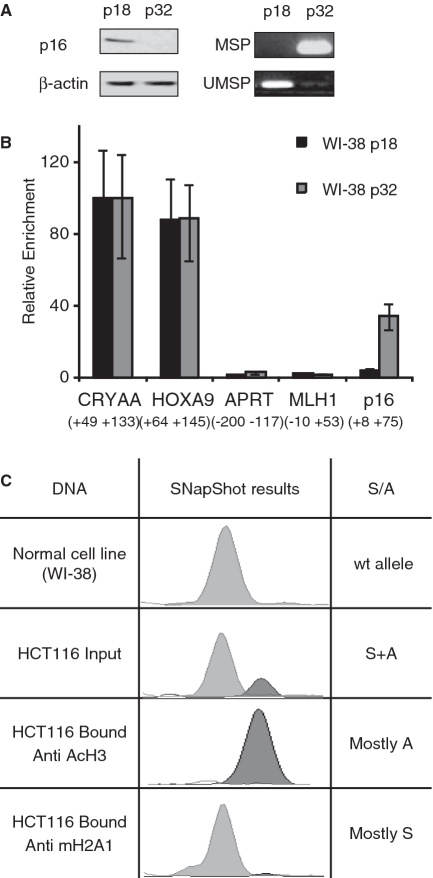
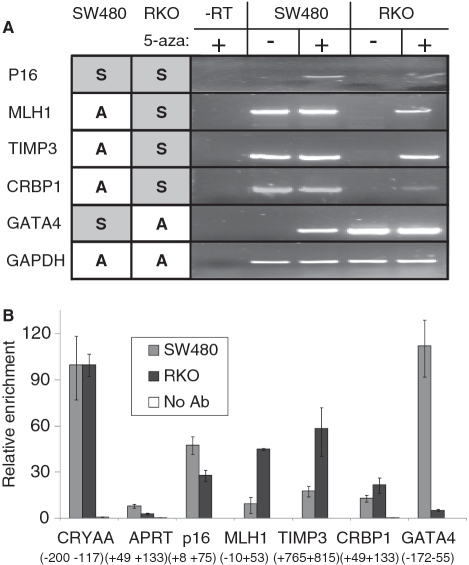
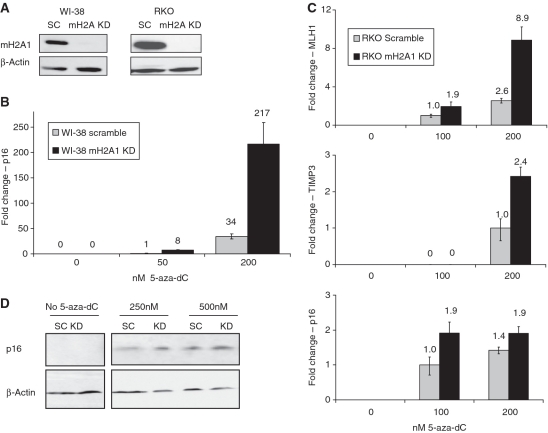
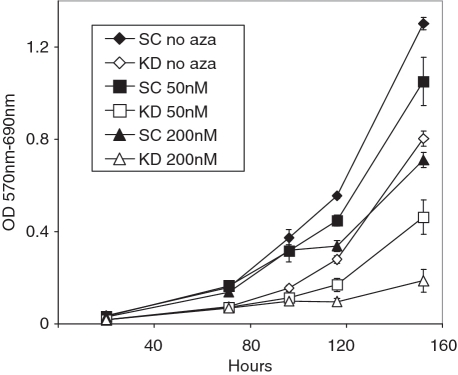
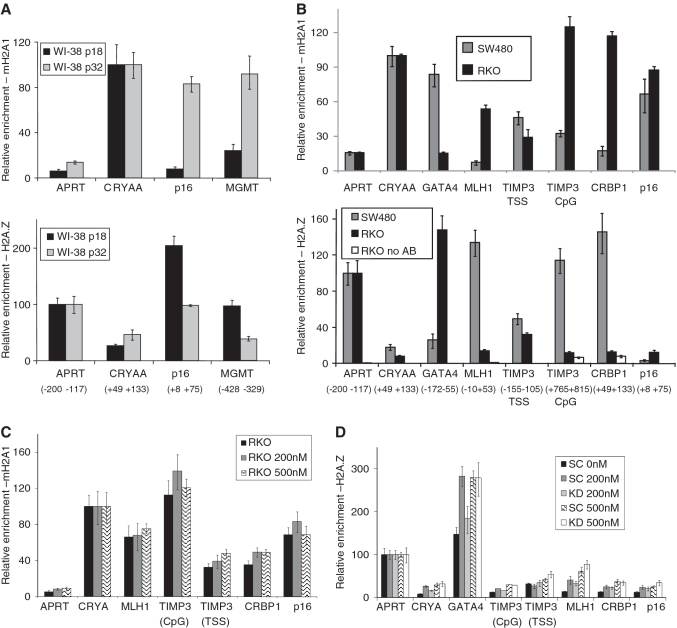
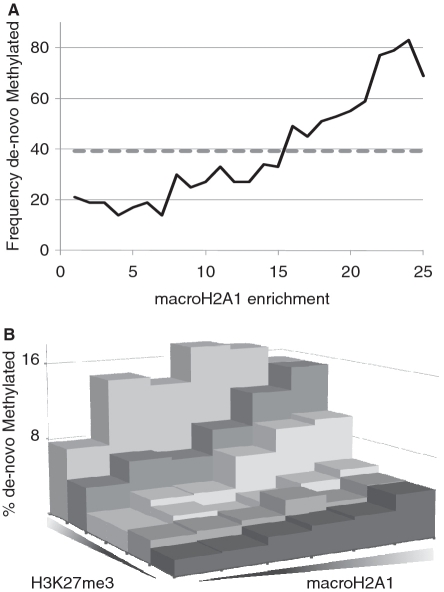
Promoter hypermethylation and heterochromatinization is a frequent event leading to gene inactivation and tumorigenesis. At the molecular level, inactivation of tumor suppressor genes in cancer has many similarities to the inactive X chromosome in female cells and is defined and maintained by DNA methylation and characteristic histone modifications. In addition, the inactive-X is marked by the histone macroH2A, a variant of H2A with a large non-histone region of unknown function. Studying tumor suppressor genes (TSGs) silenced in cancer cell lines, we find that when active, these promoters are associated with H2A.Z but become enriched for macroH2A1 once silenced. Knockdown of macroH2A1 was not sufficient for reactivation of silenced genes. However, when combined with DNA demethylation, macroH2A1 deficiency significantly enhanced reactivation of the tumor suppressor genes p16, MLH1 and Timp3 and inhibited cell proliferation. Our findings link macroH2A1 to heterochromatin of epigenetically silenced cancer genes and indicate synergism between macroH2A1 and DNA methylation in maintenance of the silenced state.
Figures
References
-
- Esteller M. Epigenetics in cancer. N. Engl. J. Med. 2008;358:1148–1159. - PubMed
-
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007;8:286–298. - PubMed
-
- Heard E. Delving into the diversity of facultative heterochromatin: the epigenetics of the inactive X chromosome. Curr. Opin. Genet. Dev. 2005;15:482–489. - PubMed
-
- Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science. 1981;211:393–396. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
