

Proteogenomic analysis of polymorphisms and gene annotation divergences in prokaryotes using a clustered mass spectrometry-friendly database
- PMID: 21030493
- PMCID: PMC3013451
- DOI: 10.1074/mcp.M110.002527
Proteogenomic analysis of polymorphisms and gene annotation divergences in prokaryotes using a clustered mass spectrometry-friendly database
Abstract
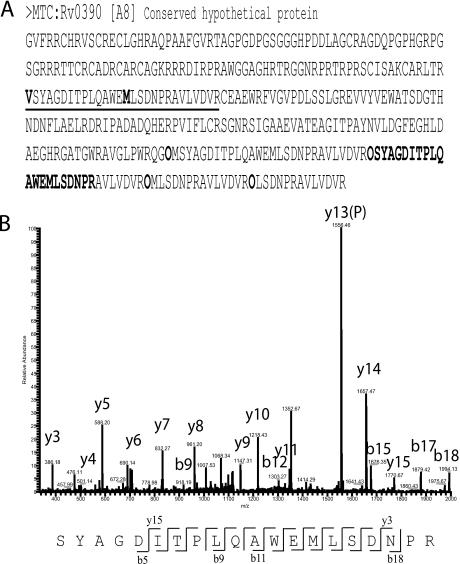
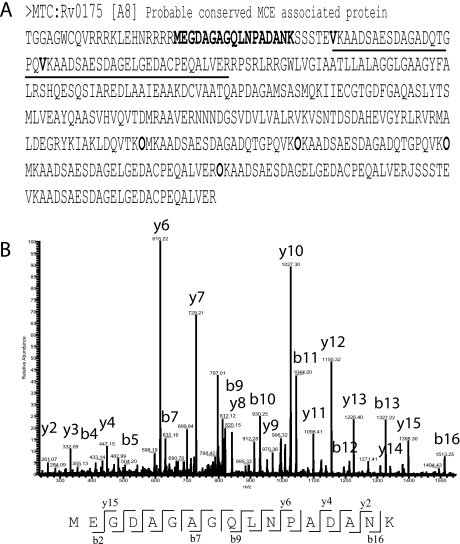
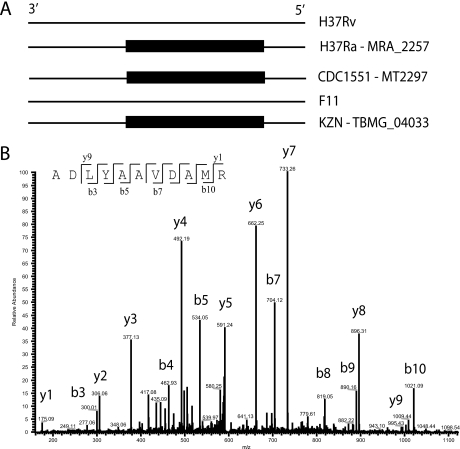
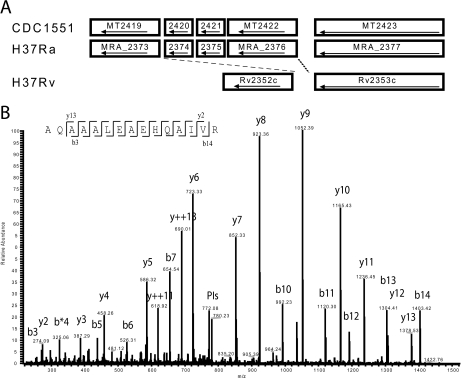
Precise annotation of genes or open reading frames is still a difficult task that results in divergence even for data generated from the same genomic sequence. This has an impact in further proteomic studies, and also compromises the characterization of clinical isolates with many specific genetic variations that may not be represented in the selected database. We recently developed software called multistrain mass spectrometry prokaryotic database builder (MSMSpdbb) that can merge protein databases from several sources and be applied on any prokaryotic organism, in a proteomic-friendly approach. We generated a database for the Mycobacterium tuberculosis complex (using three strains of Mycobacterium bovis and five of M. tuberculosis), and analyzed data collected from two laboratory strains and two clinical isolates of M. tuberculosis. We identified 2561 proteins, of which 24 were present in M. tuberculosis H37Rv samples, but not annotated in the M. tuberculosis H37Rv genome. We were also able to identify 280 nonsynonymous single amino acid polymorphisms and confirm 367 translational start sites. As a proof of concept we applied the database to whole-genome DNA sequencing data of one of the clinical isolates, which allowed the validation of 116 predicted single amino acid polymorphisms and the annotation of 131 N-terminal start sites. Moreover we identified regions not present in the original M. tuberculosis H37Rv sequence, indicating strain divergence or errors in the reference sequence. In conclusion, we demonstrated the potential of using a merged database to better characterize laboratory or clinical bacterial strains.
Figures
Similar articles
-
Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry.Mol Cell Proteomics. 2011 Dec;10(12):M111.011627. doi: 10.1074/mcp.M111.011445. Epub 2011 Oct 3. Mol Cell Proteomics. 2011. PMID: 21969609 Free PMC article.
-
Proteogenomic analysis of Mycobacterium tuberculosis Beijing B0/W148 cluster strains.J Proteomics. 2019 Feb 10;192:18-26. doi: 10.1016/j.jprot.2018.07.002. Epub 2018 Jul 24. J Proteomics. 2019. PMID: 30009986
-
Reannotation of translational start sites in the genome of Mycobacterium tuberculosis.Tuberculosis (Edinb). 2013 Jan;93(1):18-25. doi: 10.1016/j.tube.2012.11.012. Epub 2012 Dec 26. Tuberculosis (Edinb). 2013. PMID: 23273318 Free PMC article.
-
Overview of errors in the reference sequence and annotation of Mycobacterium tuberculosis H37Rv, and variation amongst its isolates.Infect Genet Evol. 2012 Jun;12(4):807-10. doi: 10.1016/j.meegid.2011.06.011. Epub 2011 Jun 23. Infect Genet Evol. 2012. PMID: 21723422 Review.
-
Limitations of the Mycobacterium tuberculosis reference genome H37Rv in the detection of virulence-related loci.Genomics. 2017 Oct;109(5-6):471-474. doi: 10.1016/j.ygeno.2017.07.004. Epub 2017 Jul 22. Genomics. 2017. PMID: 28743540 Review.
Cited by
-
Mycobacterium tuberculosis Rv3628 drives Th1-type T cell immunity via TLR2-mediated activation of dendritic cells and displays vaccine potential against the hyper-virulent Beijing K strain.Oncotarget. 2016 May 3;7(18):24962-82. doi: 10.18632/oncotarget.8771. Oncotarget. 2016. PMID: 27097115 Free PMC article.
-
Genome annotation improvements from cross-phyla proteogenomics and time-of-day differences in malaria mosquito proteins using untargeted quantitative proteomics.PLoS One. 2019 Jul 29;14(7):e0220225. doi: 10.1371/journal.pone.0220225. eCollection 2019. PLoS One. 2019. PMID: 31356616 Free PMC article.
-
Mycobacterium tuberculosis Rv0927c Inhibits NF-κB Pathway by Downregulating the Phosphorylation Level of IκBα and Enhances Mycobacterial Survival.Front Immunol. 2021 Aug 31;12:721370. doi: 10.3389/fimmu.2021.721370. eCollection 2021. Front Immunol. 2021. PMID: 34531869 Free PMC article.
-
Introducing the ESAT-6 free IGRA, a companion diagnostic for TB vaccines based on ESAT-6.Sci Rep. 2017 Apr 7;7:45969. doi: 10.1038/srep45969. Sci Rep. 2017. PMID: 28387329 Free PMC article.
-
Empowering Shotgun Mass Spectrometry with 2DE: A HepG2 Study.Int J Mol Sci. 2020 May 27;21(11):3813. doi: 10.3390/ijms21113813. Int J Mol Sci. 2020. PMID: 32471280 Free PMC article.
References
-
- Garrels J. I. (2002) Yeast genomic databases and the challenge of the post-genomic era. Funct. Integr. Genomics 2, 212–237 - PubMed
-
- Rappsilber J., Mann M. (2002) What does it mean to identify a protein in proteomics? Trends Biochem. Sci. 27, 74–78 - PubMed
-
- Ge H., Walhout A. J., Vidal M. (2003) Integrating ‘omic’ information: a bridge between genomics and systems biology. Trends. Genet. 19, 551–560 - PubMed
-
- Kyrpides N. C. (1999) Genomes OnLine Database (GOLD 1.0): a monitor of complete and ongoing genome projects world-wide. Bioinformatics 15, 773–774 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
