

The nucleotide addition cycle of RNA polymerase is controlled by two molecular hinges in the Bridge Helix domain
- PMID: 21034443
- PMCID: PMC2988716
- DOI: 10.1186/1741-7007-8-134
The nucleotide addition cycle of RNA polymerase is controlled by two molecular hinges in the Bridge Helix domain
Abstract
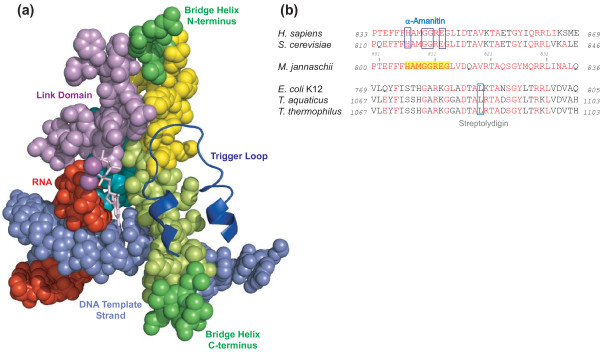
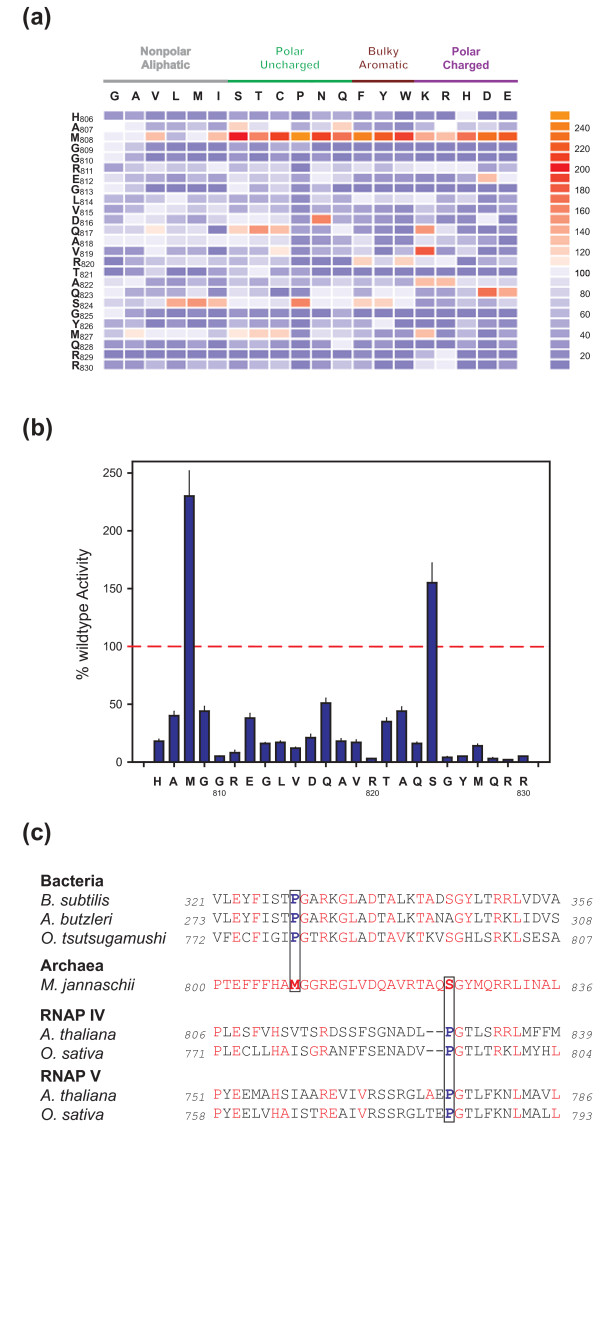
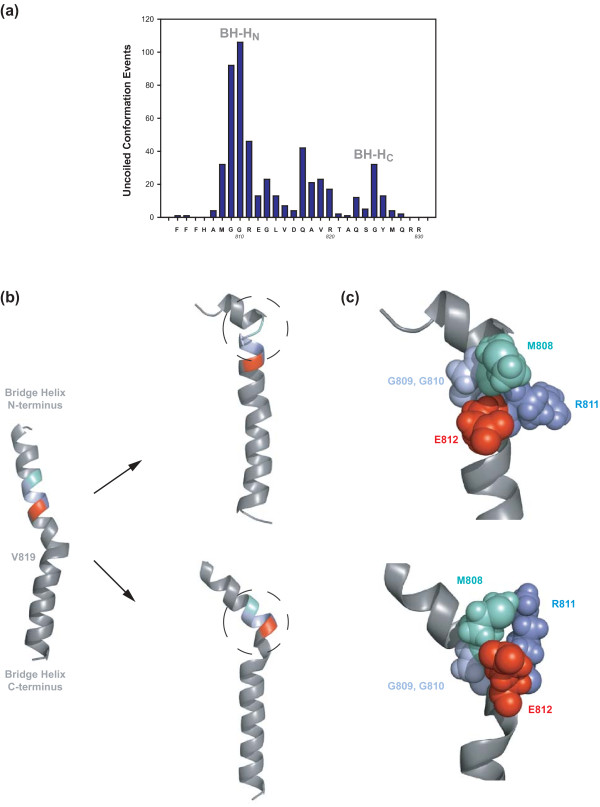
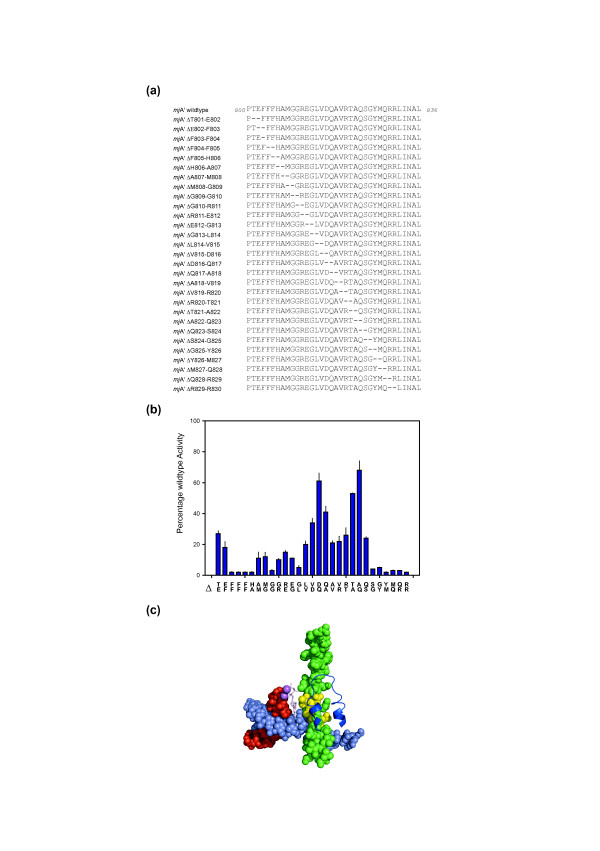
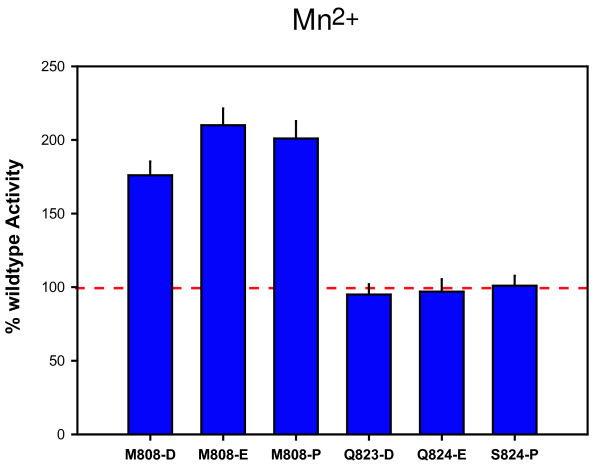
Background: Cellular RNA polymerases (RNAPs) are complex molecular machines that combine catalysis with concerted conformational changes in the active center. Previous work showed that kinking of a hinge region near the C-terminus of the Bridge Helix (BH-H(C)) plays a critical role in controlling the catalytic rate.
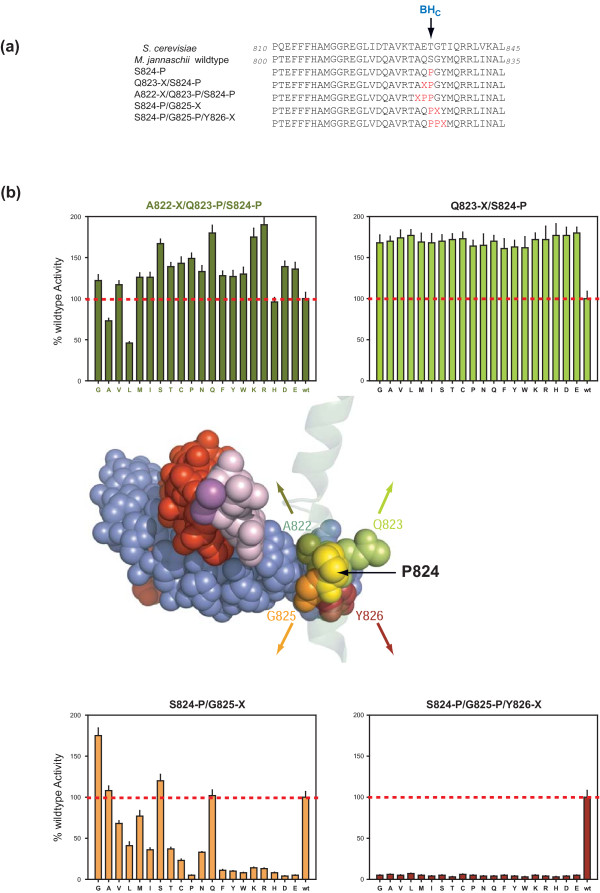
Results: Here, new evidence for the existence of an additional hinge region in the amino-terminal portion of the Bridge Helix domain (BH-H(N)) is presented. The nanomechanical properties of BH-H(N) emerge as a direct consequence of the highly conserved primary amino acid sequence. Mutations that are predicted to influence its flexibility cause corresponding changes in the rate of the nucleotide addition cycle (NAC). BH-H(N) displays functional properties that are distinct from BH-H(C), suggesting that conformational changes in the Bridge Helix control the NAC via two independent mechanisms.
Conclusions: The properties of two distinct molecular hinges in the Bridge Helix of RNAP determine the functional contribution of this domain to key stages of the NAC by coordinating conformational changes in surrounding domains.
Figures
Similar articles
-
Cation-π interactions induce kinking of a molecular hinge in the RNA polymerase bridge-helix domain.Biochem Soc Trans. 2011 Jan;39(1):31-5. doi: 10.1042/BST0390031. Biochem Soc Trans. 2011. PMID: 21265743 Review.
-
The Bridge Helix of RNA polymerase acts as a central nanomechanical switchboard for coordinating catalysis and substrate movement.Archaea. 2011;2011:608385. doi: 10.1155/2011/608385. Epub 2012 Jan 22. Archaea. 2011. PMID: 22312317 Free PMC article. Review.
-
The bridge helix coordinates movements of modules in RNA polymerase.BMC Biol. 2010 Nov 29;8:141. doi: 10.1186/1741-7007-8-141. BMC Biol. 2010. PMID: 21114873 Free PMC article.
-
The RNA polymerase bridge helix YFI motif in catalysis, fidelity and translocation.Biochim Biophys Acta. 2013 Feb;1829(2):187-98. doi: 10.1016/j.bbagrm.2012.11.005. Epub 2012 Nov 30. Biochim Biophys Acta. 2013. PMID: 23202476 Free PMC article.
-
Activity map of the Escherichia coli RNA polymerase bridge helix.J Biol Chem. 2011 Apr 22;286(16):14469-79. doi: 10.1074/jbc.M110.212902. Epub 2011 Feb 25. J Biol Chem. 2011. PMID: 21357417 Free PMC article.
Cited by
-
Computational simulation strategies for analysis of multisubunit RNA polymerases.Chem Rev. 2013 Nov 13;113(11):8546-66. doi: 10.1021/cr400046x. Epub 2013 Aug 29. Chem Rev. 2013. PMID: 23987500 Free PMC article. Review. No abstract available.
-
CBR antimicrobials inhibit RNA polymerase via at least two bridge-helix cap-mediated effects on nucleotide addition.Proc Natl Acad Sci U S A. 2015 Aug 4;112(31):E4178-87. doi: 10.1073/pnas.1502368112. Epub 2015 Jul 20. Proc Natl Acad Sci U S A. 2015. PMID: 26195788 Free PMC article.
-
Functional assays for transcription mechanisms in high-throughput.Methods. 2019 Apr 15;159-160:115-123. doi: 10.1016/j.ymeth.2019.02.017. Epub 2019 Feb 20. Methods. 2019. PMID: 30797033 Free PMC article. Review.
-
Trigger-helix folding pathway and SI3 mediate catalysis and hairpin-stabilized pausing by Escherichia coli RNA polymerase.Nucleic Acids Res. 2014 Nov 10;42(20):12707-21. doi: 10.1093/nar/gku997. Epub 2014 Oct 21. Nucleic Acids Res. 2014. PMID: 25336618 Free PMC article.
-
Transcription elongation of the plant RNA polymerase IV is prone to backtracking.Sci Adv. 2024 Aug 23;10(34):eadq3087. doi: 10.1126/sciadv.adq3087. Epub 2024 Aug 23. Sci Adv. 2024. PMID: 39178250 Free PMC article.
References
-
- Kireeva M, Kashlev M, Burton ZF. Translocation by multi-subunit RNA polymerases. Biochim Biophys Acta. 2010;1799:389–401. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
