

Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci
- PMID: 21035756
- PMCID: PMC3026436
- DOI: 10.1016/j.cmet.2010.09.012
Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci
Erratum in
- Cell Metab. 2010 Dec 1;12(6):683
Abstract
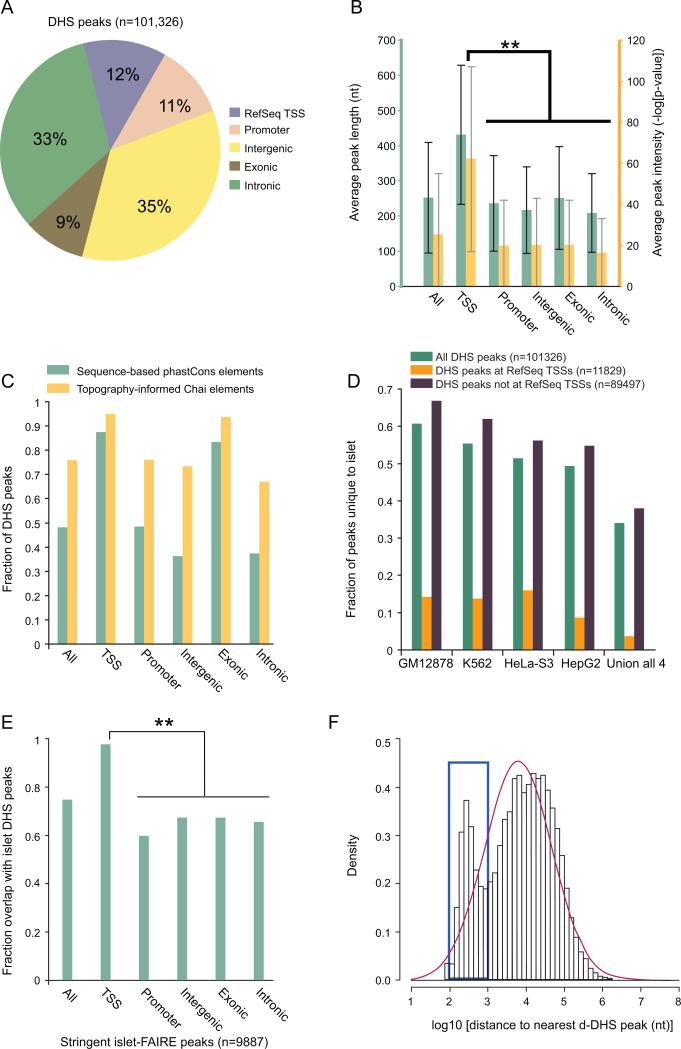
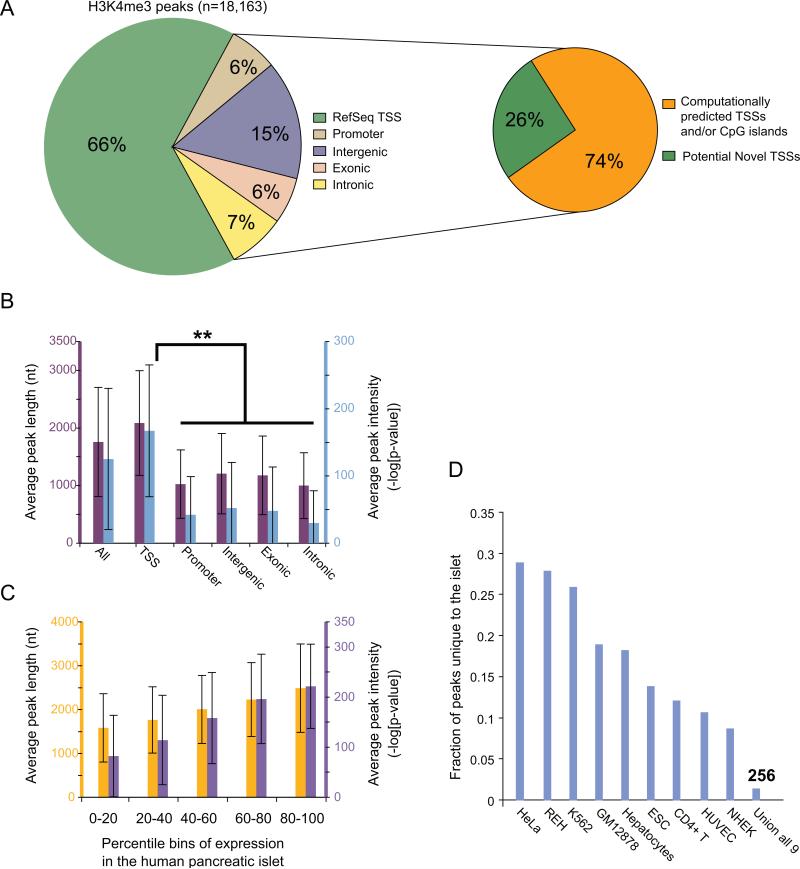
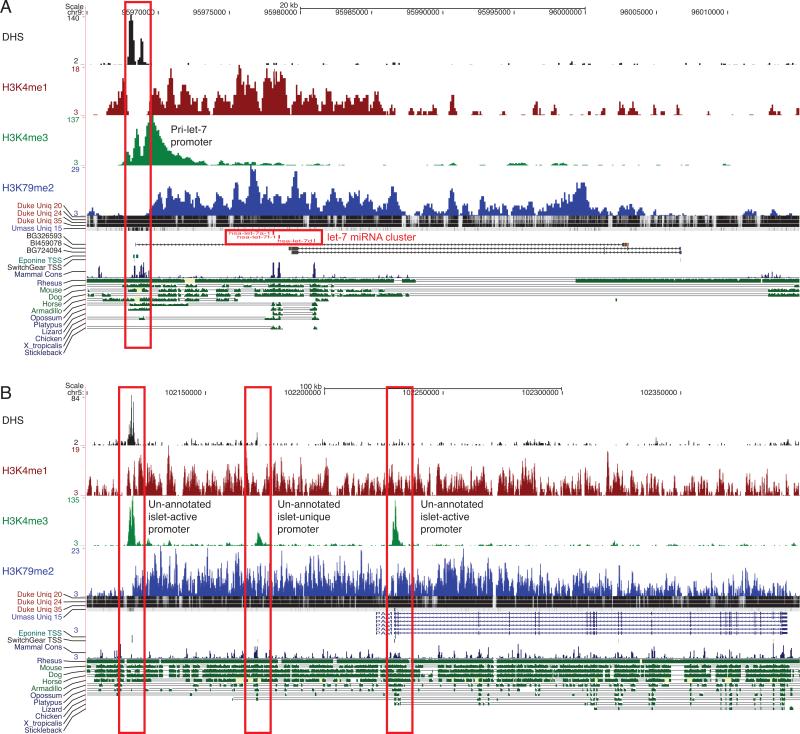
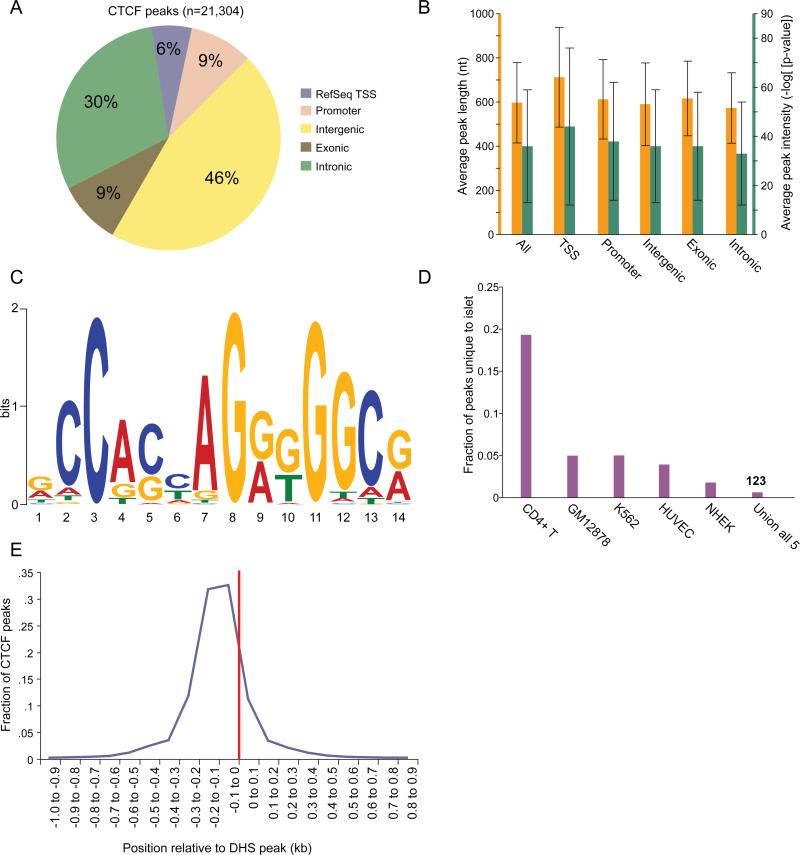
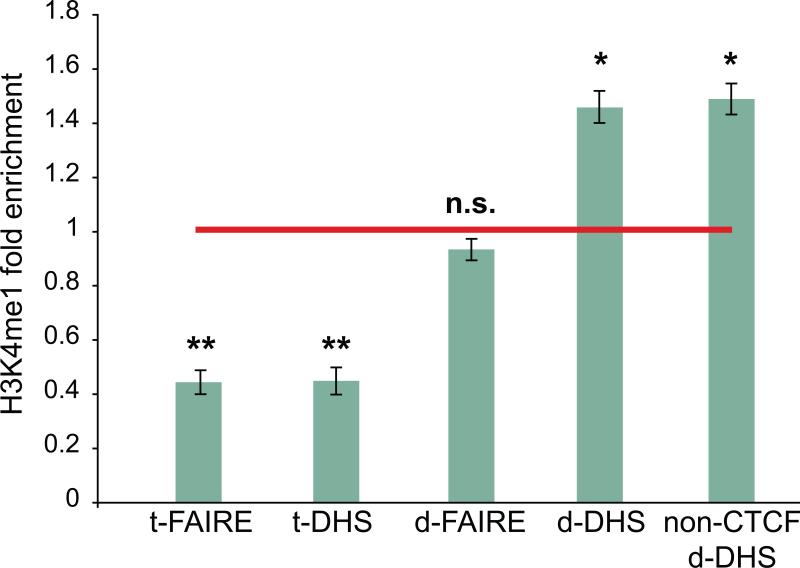
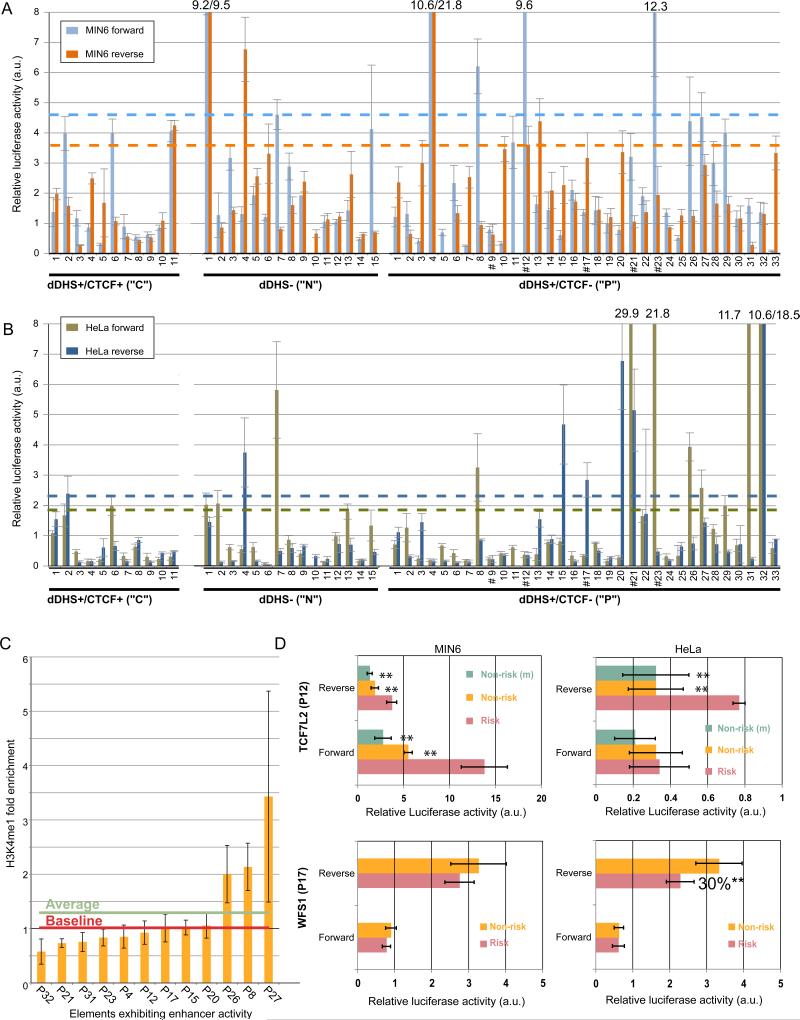
Identifying cis-regulatory elements is important to understanding how human pancreatic islets modulate gene expression in physiologic or pathophysiologic (e.g., diabetic) conditions. We conducted genome-wide analysis of DNase I hypersensitive sites, histone H3 lysine methylation modifications (K4me1, K4me3, K79me2), and CCCTC factor (CTCF) binding in human islets. This identified ∼18,000 putative promoters (several hundred unannotated and islet-active). Surprisingly, active promoter modifications were absent at genes encoding islet-specific hormones, suggesting a distinct regulatory mechanism. Of 34,039 distal (nonpromoter) regulatory elements, 47% are islet unique and 22% are CTCF bound. In the 18 type 2 diabetes (T2D)-associated loci, we identified 118 putative regulatory elements and confirmed enhancer activity for 12 of 33 tested. Among six regulatory elements harboring T2D-associated variants, two exhibit significant allele-specific differences in activity. These findings present a global snapshot of the human islet epigenome and should provide functional context for noncoding variants emerging from genetic studies of T2D and other islet disorders.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. - PubMed
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
