

Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies
- PMID: 21036400
- PMCID: PMC3044822
- DOI: 10.1016/j.ophtha.2010.07.029
Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies
Abstract
Purpose: Autosomal-dominant optic atrophy (DOA) is one of the most common inherited optic neuropathies, and it is genetically heterogeneous, with mutations in both OPA1 and OPA3 known to cause disease. Approximately 60% of cases harbor OPA1 mutations, whereas OPA3 mutations have been reported in only 2 pedigrees with DOA and premature cataracts. The aim of this study was to determine the yield of OPA1 and OPA3 screening in a cohort of presumed DOA cases referred to a tertiary diagnostic laboratory.
Design: Retrospective case series.
Participants: One hundred eighty-eight probands with bilateral optic atrophy referred for molecular genetic investigations at a tertiary diagnostic facility: 38 patients with an autosomal-dominant pattern of inheritance and 150 sporadic cases.
Methods: OPA1 and OPA3 genetic testing was initially performed using polymerase chain reaction-based sequencing methods. The presence of large-scale OPA1 and OPA3 genomic rearrangements was assessed further with a targeted comparative genomic hybridization microarray platform. The 3 primary Leber hereditary optic neuropathy (LHON) mutations, m.3460G→>A, m.11778G→A, and m.14484T→C, also were screened in all patients.
Main outcome measures: The proportion of patients with OPA1 and OPA3 pathogenic mutations. The clinical profile observed in molecularly confirmed DOA cases.
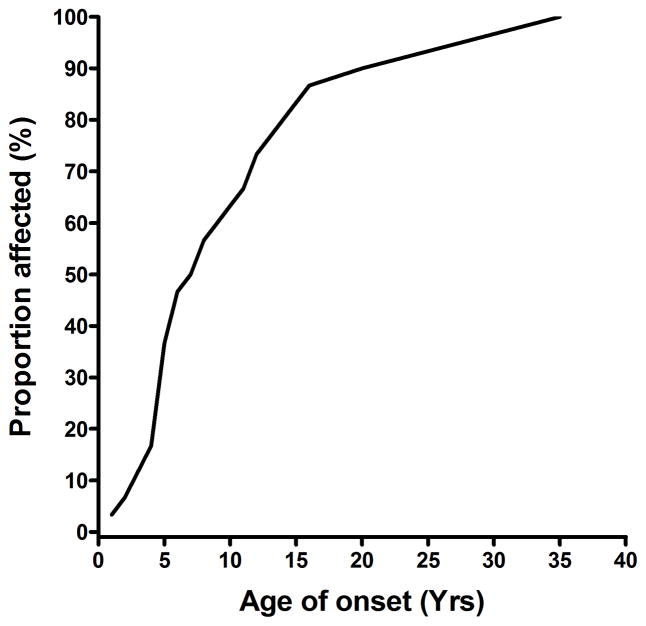
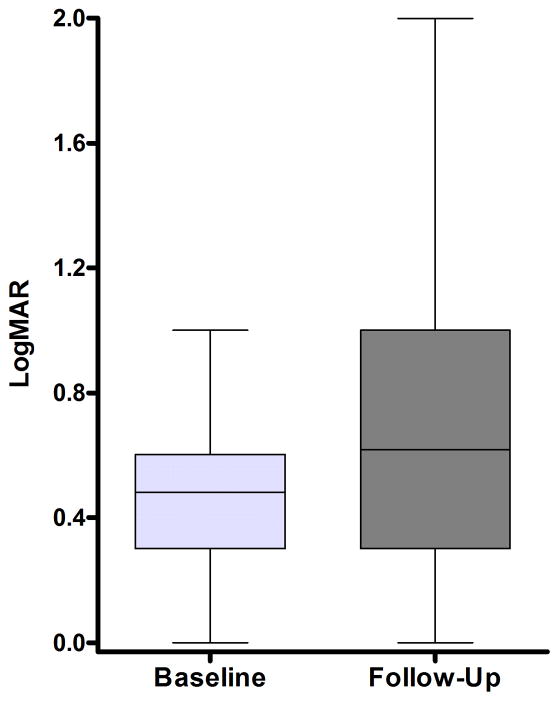
Results: Twenty-one different OPA1 mutations were found in 27 (14.4%) of the 188 probands screened. The mutations included 6 novel pathogenic variants and the first reported OPA1 initiation codon mutation at c.1A→T. An OPA1 missense mutation, c.239A→G (p.Y80C), was identified in an 11-year-old black girl with optic atrophy and peripheral sensorimotor neuropathy in her lower limbs. The OPA1 detection rate was significantly higher among individuals with a positive family history of visual failure (50.0%) compared with sporadic cases (5.3%). The primary LHON screen was negative in the patient cohort, and additional molecular investigations did not reveal any large-scale OPA1 rearrangements or OPA3 genetic defects. The mean baseline visual acuity for the OPA1-positive group was 0.48 logarithm of the minimum angle of resolution (units mean Snellen equivalent, 20/61; range, 20/20-20/400; 95% confidence interval, 20/52-20/71), and visual deterioration occurred in 54.2% of patients during follow-up.
Conclusions: OPA1 mutations are the most common genetic defects identified in patients with suspected DOA, whereas OPA3 mutations are very rare in isolated optic atrophy cases.
Copyright © 2011 American Academy of Ophthalmology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Newman NJ, Biousse V. Hereditary optic neuropathies. Eye (Lond) 2004;18:1144–60. - PubMed
-
- Votruba M. Molecular genetic basis of primary inherited optic neuropathies. Eye (Lond) 2004;18:1126–32. - PubMed
-
- Thiselton DL, Alexander C, Taanman JW, et al. A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. 2002;43:1715–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
