

Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity
- PMID: 21037571
- PMCID: PMC2999892
- DOI: 10.1038/ng.705
Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity
Abstract
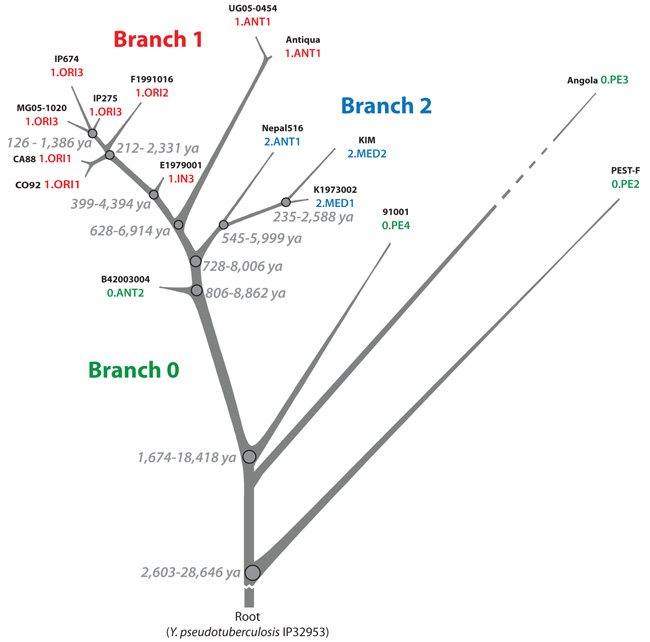
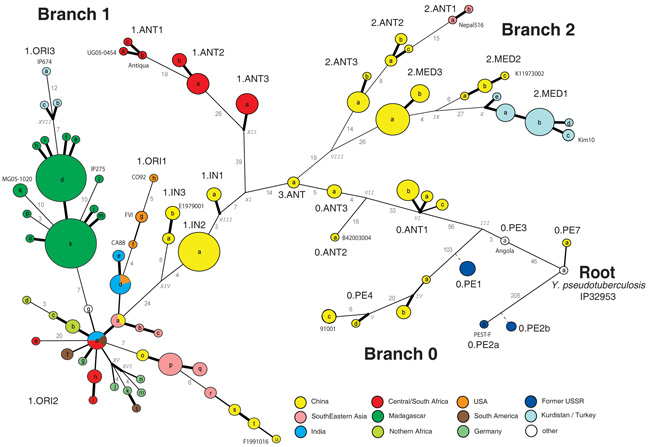
Plague is a pandemic human invasive disease caused by the bacterial agent Yersinia pestis. We here report a comparison of 17 whole genomes of Y. pestis isolates from global sources. We also screened a global collection of 286 Y. pestis isolates for 933 SNPs using Sequenom MassArray SNP typing. We conducted phylogenetic analyses on this sequence variation dataset, assigned isolates to populations based on maximum parsimony and, from these results, made inferences regarding historical transmission routes. Our phylogenetic analysis suggests that Y. pestis evolved in or near China and spread through multiple radiations to Europe, South America, Africa and Southeast Asia, leading to country-specific lineages that can be traced by lineage-specific SNPs. All 626 current isolates from the United States reflect one radiation, and 82 isolates from Madagascar represent a second radiation. Subsequent local microevolution of Y. pestis is marked by sequential, geographically specific SNPs.
Figures
References
-
- Diamond J. Guns, germs and steel. Jonathan Cape; London: 1997. pp. 1–480.
-
- Keeling MJ, Gilligan CA. Metapopulation dynamics of bubonic plague. Nature. 2000;407:903–906. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
