

Phosphatidylserine as an anchor for plasminogen and its plasminogen receptor, histone H2B, to the macrophage surface
- PMID: 21040449
- PMCID: PMC3881192
- DOI: 10.1111/j.1538-7836.2010.04132.x
Phosphatidylserine as an anchor for plasminogen and its plasminogen receptor, histone H2B, to the macrophage surface
Abstract
Background: Plasminogen (Plg) binding to cell surface Plg receptors (Plg-Rs) on the surface of macrophages facilitates Plg activation and migration of these cells. Histone H2B (H2B) acts as a Plg-R and its cell surface expression is up-regulated when monocytes are differentiated to macrophages via a pathway dependent on L-type Ca(2+) channels and intracellular Ca(2+).
Objectives: We sought to investigate the mechanism by which H2B, a protein without a transmembrane domain, is retained on the macrophage surface.
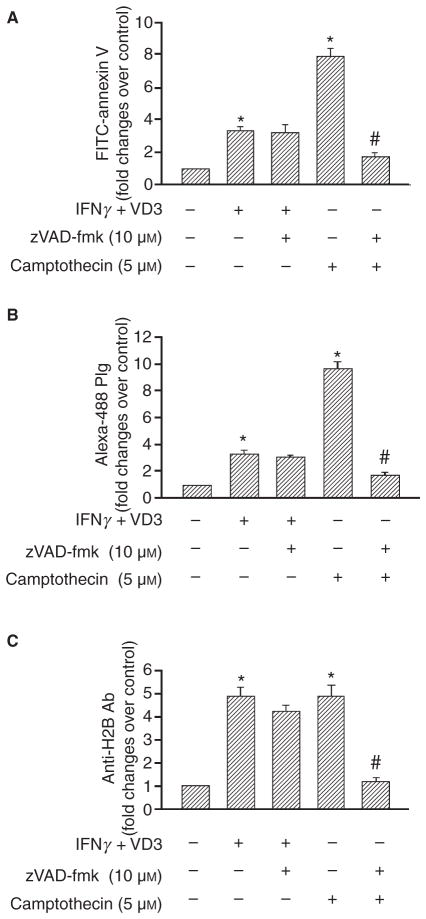
Methods: THP-1 monocytoid cells were induced to differentiate with interferon gamma + Vitamin D3 or to undergo apoptosis by treatment with camptothecin. Flow cytometry and cell surface biotinylation followed by Western blotting were used to measure the interrelationship between Plg binding, cell surface expression of H2B and outer membrane exposure of phosphatidylserine (PS).
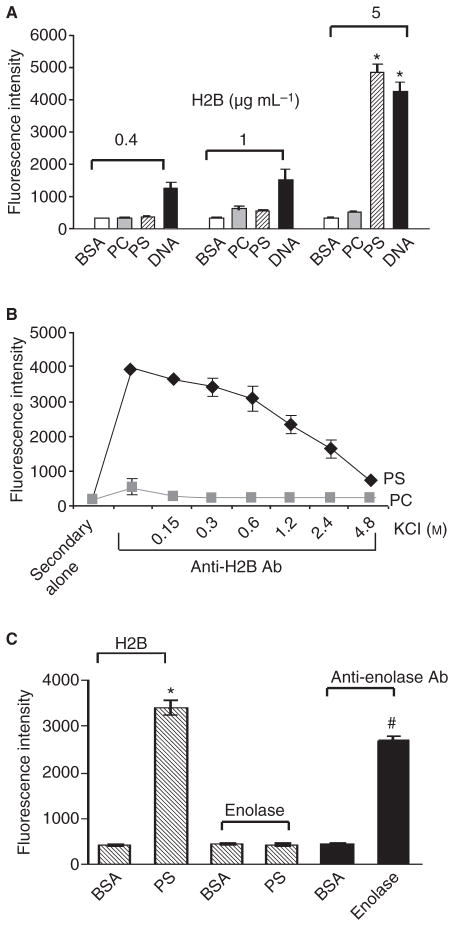
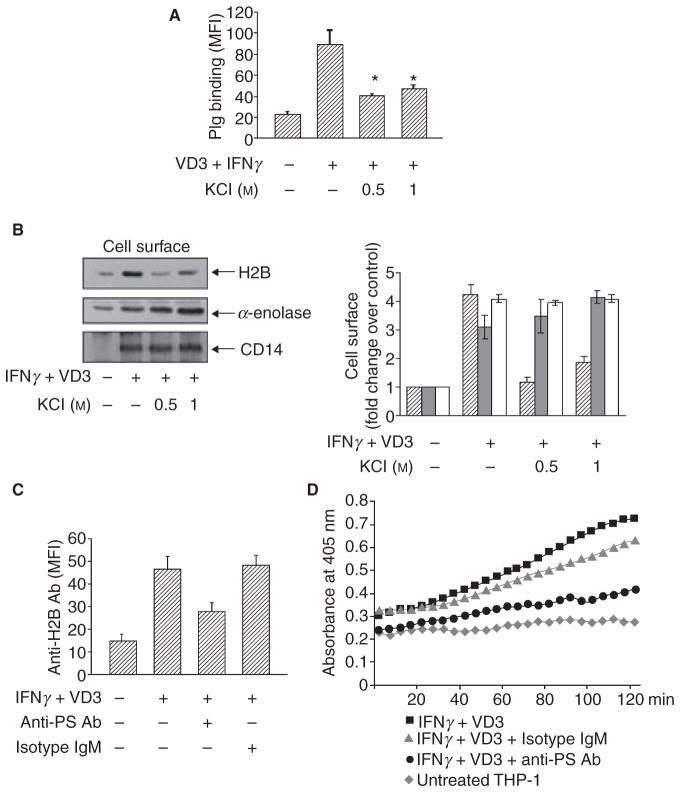
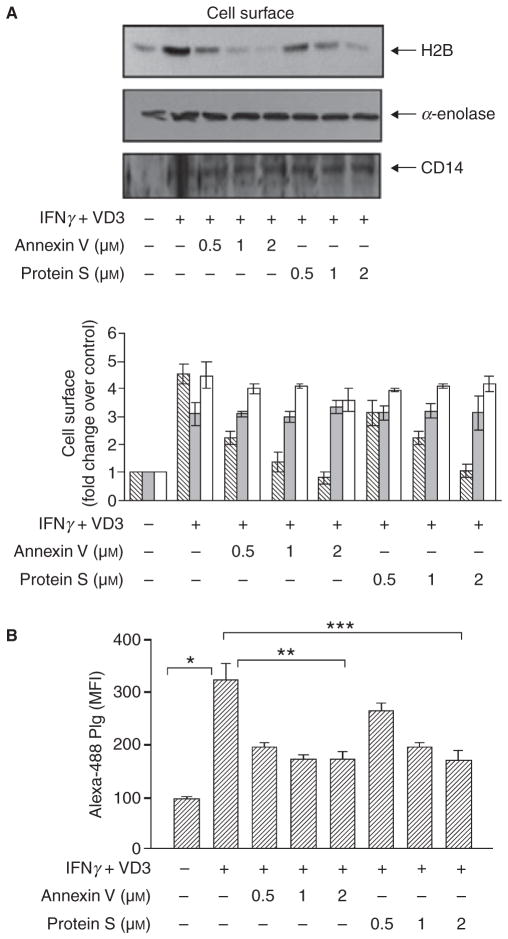
Results: H2B interacted directly with PS via an electrostatic interaction. Anti-PS or PS binding proteins, annexin V and protein S, diminished H2B interaction with PS on the surface of differentiated or apoptotic cells and these same reagents inhibited Plg binding to these cells. L-type Ca(2+) channels played a significant role in PS exposure, H2B surface expression and Plg binding induced either by differentiation or apoptosis.
Conclusions: These data suggest that H2B tethers to the surface of cells by interacting with PS on differentiated or apoptotic monocytoid cells. L-type Ca(2+) channels regulate PS exposure on the surface of these cells. The exposed PS interacts directly with H2B and hence provides sites for Plg to bind to.
© 2011 International Society on Thrombosis and Haemostasis.
Conflict of interest statement
The authors state that they have no conflict of interest.
Figures
Similar articles
-
Histone H2B as a functionally important plasminogen receptor on macrophages.Blood. 2007 Nov 15;110(10):3763-72. doi: 10.1182/blood-2007-03-079392. Epub 2007 Aug 9. Blood. 2007. PMID: 17690254 Free PMC article.
-
Plasminogen-induced foam cell formation by macrophages occurs through a histone 2B (H2B)-PAR1 pathway and requires integrity of clathrin-coated pits.J Thromb Haemost. 2021 Apr;19(4):941-953. doi: 10.1111/jth.15253. Epub 2021 Feb 17. J Thromb Haemost. 2021. PMID: 33492784
-
L-type calcium channel blockers exert an antiinflammatory effect by suppressing expression of plasminogen receptors on macrophages.Circ Res. 2009 Jul 17;105(2):167-75. doi: 10.1161/CIRCRESAHA.109.200311. Epub 2009 Jun 11. Circ Res. 2009. PMID: 19520970 Free PMC article.
-
New insights into the role of Plg-RKT in macrophage recruitment.Int Rev Cell Mol Biol. 2014;309:259-302. doi: 10.1016/B978-0-12-800255-1.00005-3. Int Rev Cell Mol Biol. 2014. PMID: 24529725 Free PMC article. Review.
-
The plasminogen receptor, Plg-R(KT), and macrophage function.J Biomed Biotechnol. 2012;2012:250464. doi: 10.1155/2012/250464. Epub 2012 Oct 14. J Biomed Biotechnol. 2012. PMID: 23125524 Free PMC article. Review.
Cited by
-
From plasminogen to plasmin: role of plasminogen receptors in human cancer.Int J Mol Sci. 2014 Nov 17;15(11):21229-52. doi: 10.3390/ijms151121229. Int J Mol Sci. 2014. PMID: 25407528 Free PMC article. Review.
-
So many plasminogen receptors: why?J Biomed Biotechnol. 2012;2012:141806. doi: 10.1155/2012/141806. Epub 2012 Oct 14. J Biomed Biotechnol. 2012. PMID: 23118495 Free PMC article. Review.
-
A novel regulatory pathway for autoimmune disease: binding of partial MHC class II constructs to monocytes reduces CD74 expression and induces both specific and bystander T-cell tolerance.J Autoimmun. 2013 Feb;40:96-110. doi: 10.1016/j.jaut.2012.08.004. Epub 2012 Sep 29. J Autoimmun. 2013. PMID: 23026773 Free PMC article.
-
Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion.EBioMedicine. 2020 Mar;53:102671. doi: 10.1016/j.ebiom.2020.102671. Epub 2020 Feb 27. EBioMedicine. 2020. PMID: 32114386 Free PMC article.
-
Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice.Sci Rep. 2018 Mar 5;8(1):4020. doi: 10.1038/s41598-018-22156-5. Sci Rep. 2018. PMID: 29507382 Free PMC article.
References
-
- Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. The cell biology of the plasminogen system. FASEB J. 1995;9:939–45. - PubMed
-
- Miles LA, Hawley SB, Baik N, Andronicos NM, Castellino FJ, Parmer RJ. Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front Biosci. 2005;10:1754–62. - PubMed
-
- Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF. Role of cell-surface lysines in plasminogen binding to cells: identification of alpha-enolase as a candidate plasminogen receptor. Biochemistry. 1991;30:1682–91. - PubMed
-
- Herren T, Burke TA, Das R, Plow EF. Identification of histone H2B as a regulated plasminogen receptor. Biochemistry. 2006;45:9463–74. - PubMed
-
- Wygrecka M, Marsh LM, Morty RE, Henneke I, Guenther A, Lohmeyer J, Markart P, Preissner KT. Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood. 2009;113:5588–98. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
