

Gene expression profiling of human breast tissue samples using SAGE-Seq
- PMID: 21045080
- PMCID: PMC2989999
- DOI: 10.1101/gr.108217.110
Gene expression profiling of human breast tissue samples using SAGE-Seq
Abstract
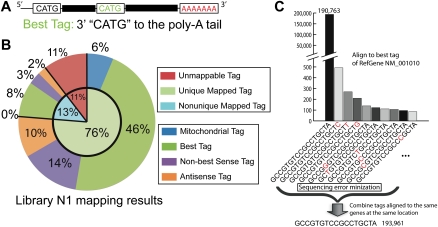
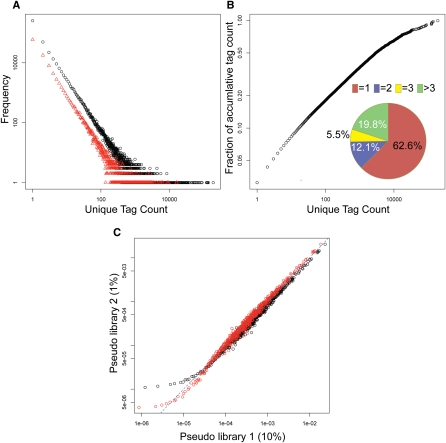
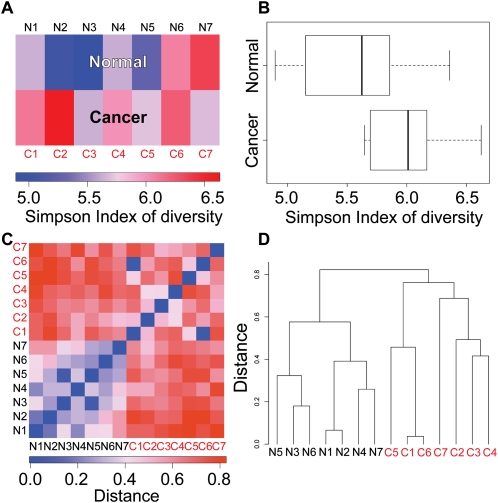
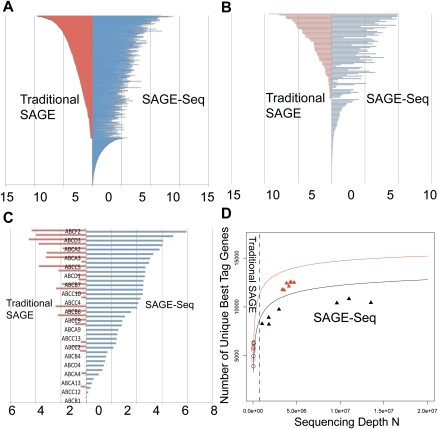
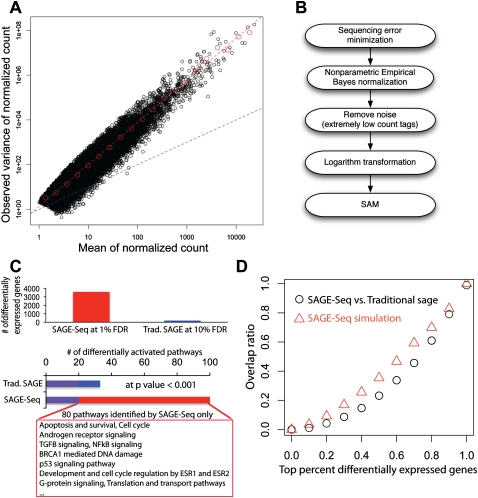
We present a powerful application of ultra high-throughput sequencing, SAGE-Seq, for the accurate quantification of normal and neoplastic mammary epithelial cell transcriptomes. We develop data analysis pipelines that allow the mapping of sense and antisense strands of mitochondrial and RefSeq genes, the normalization between libraries, and the identification of differentially expressed genes. We find that the diversity of cancer transcriptomes is significantly higher than that of normal cells. Our analysis indicates that transcript discovery plateaus at 10 million reads/sample, and suggests a minimum desired sequencing depth around five million reads. Comparison of SAGE-Seq and traditional SAGE on normal and cancerous breast tissues reveals higher sensitivity of SAGE-Seq to detect less-abundant genes, including those encoding for known breast cancer-related transcription factors and G protein-coupled receptors (GPCRs). SAGE-Seq is able to identify genes and pathways abnormally activated in breast cancer that traditional SAGE failed to call. SAGE-Seq is a powerful method for the identification of biomarkers and therapeutic targets in human disease.
Figures
References
-
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, et al. 1991. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 252: 1651–1656 - PubMed
-
- Baggerly KA, Deng L, Morris JS, Aldaz CM 2003. Differential expression in SAGE: Accounting for normal between-library variation. Bioinformatics 19: 1477–1483 - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous