

Cri-du-Chat Syndrome Cytogenetically Cryptic Recombination Aneusomy of Chromosome 5: Implications in Recurrence Risk Estimation
- PMID: 21045963
- PMCID: PMC2941846
- DOI: 10.1159/000319321
Cri-du-Chat Syndrome Cytogenetically Cryptic Recombination Aneusomy of Chromosome 5: Implications in Recurrence Risk Estimation
Abstract
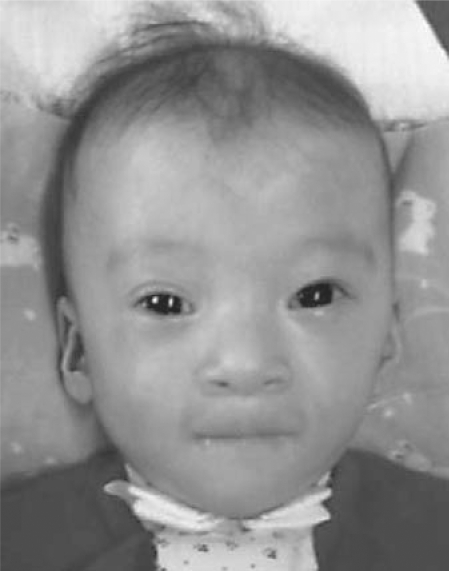
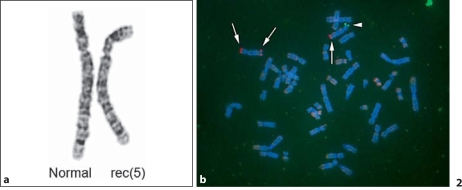
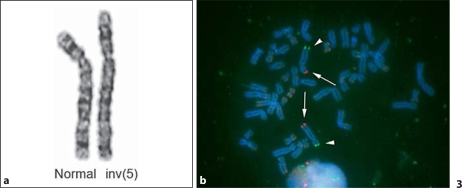
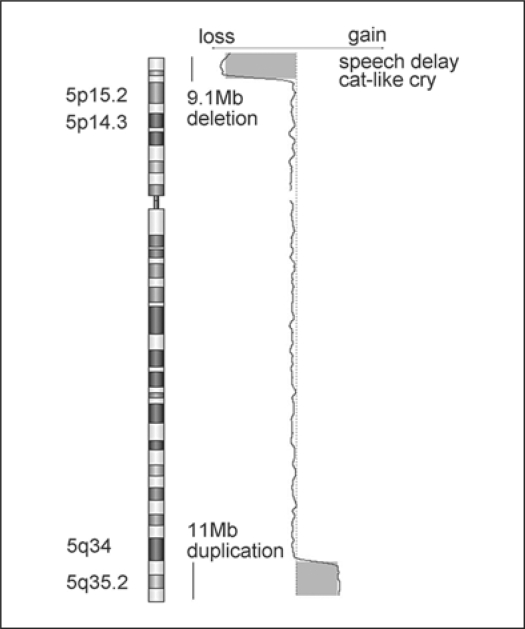
Cri-du-chat syndrome is caused by haploinsufficiency of the genes on the distal part of the short arm of chromosome 5, and characteristic features include microcephaly, developmental delays, and a distinctive high-pitched mewing cry. Most cri-du-chat syndrome cases result from a sporadic de novo deletion that is associated with a low recurrence risk. On rare occasions, however, cri-du-chat syndrome with 5p monosomy can be accompanied by 5q trisomy. This combination is virtually always associated with parental large pericentric inversions. Among previously reported cri-du-chat syndrome cases with 5p monosomy accompanied by 5q trisomy, the aneusomy of chromosome 5 in all but one case was cytogenetically visible using G-banding. When an accompanying 5q trisomy is detected, a significant recurrence risk is expected. We here report on a patient with cri-du-chat syndrome phenotype who initially exhibited a normal karyotype on G-banding but in whom molecular analysis using multiplex ligation-dependent probe amplification and array comparative genomic hybridization revealed a 5p deletion accompanied by a 5q duplication. Parental chromosomal testing led to the identification of a very large pericentric inversion, of which breakpoints resided at the terminal regions of 5p15.31 and 5q35.1. This information was vital for counseling the family regarding the significantly high recurrence risk.
Figures
References
-
- Akalin I, Yararbas K, Akgul N, Babaoglu E, Akay GG, et al. del5p/dup5q in a ‘cri du chat’ patient without parental chromosomal rearrangement. Am J Med Genet A. 2006;140:1016–1020. - PubMed
-
- Anton E, Blanco J, Egozcue J, Vidal F. Sperm studies in heterozygote inversion carriers: a review. Cytogenet Genome Res. 2005;111:297–304. - PubMed
-
- Beemer FA, de France HF, Rosina-Angelista IJ, Gerards LJ, Cats BP, et al. Familial partial monosomy 5p and trisomy 5q; three cases due to paternal pericentric inversion 5 (p151q333) Clin Genet. 1984;26:209–215. - PubMed
-
- Bocian E, Suchenek K, Obersztyn E, Nowakowska B, Mazurczak T. Recombination aneusomy of subtelomeric regions of chromosome 5, resulting from a large familial pericentric inversion inv(5)(p15.33q35.3) J Appl Genet. 2005;46:109–114. - PubMed
-
- de Perdigo A, Gabriel-Robez O, Rumpler Y. Correlation between chromosomal breakpoint positions and synaptic behaviour in human males heterozygous for a pericentric inversion. Hum Genet. 1989;83:274–276. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
