

Predicting gene function using few positive examples and unlabeled ones
- PMID: 21047378
- PMCID: PMC2975410
- DOI: 10.1186/1471-2164-11-S2-S11
Predicting gene function using few positive examples and unlabeled ones
Abstract
Background: A large amount of functional genomic data have provided enough knowledge in predicting gene function computationally, which uses known functional annotations and relationship between unknown genes and known ones to map unknown genes to GO functional terms. The prediction procedure is usually formulated as binary classification problem. Training binary classifier needs both positive examples and negative ones that have almost the same size. However, from various annotation database, we can only obtain few positive genes annotation for most of functional terms, that is, there are only few positive examples for training classifier, which makes predicting directly gene function infeasible.
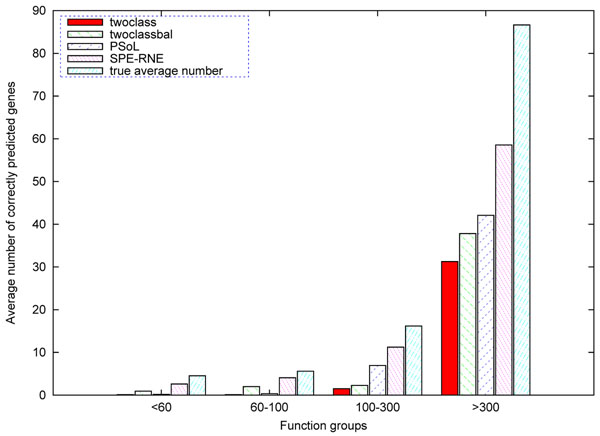
Results: We propose a novel approach SPE_RNE to train classifier for each functional term. Firstly, positive examples set is enlarged by creating synthetic positive examples. Secondly, representative negative examples are selected by training SVM (support vector machine) iteratively to move classification hyperplane to a appropriate place. Lastly, an optimal SVM classifier are trained by using grid search technique. On combined kernel of Yeast protein sequence, microarray expression, protein-protein interaction and GO functional annotation data, we compare SPE_RNE with other three typical methods in three classical performance measures recall R, precise P and their combination F: twoclass considers all unlabeled genes as negative examples, twoclassbal selects randomly same number negative examples from unlabeled gene, PSoL selects a negative examples set that are far from positive examples and far from each other.
Conclusions: In test data and unknown genes data, we compute average and variant of measure F. The experiments show that our approach has better generalized performance and practical prediction capacity. In addition, our method can also be used for other organisms such as human.
Figures


 and initial negative example set N0,the SVM classifier C0 is learned, the negative example set N1 consists of the support vectors of C0 and the unlabeled examples predicted as negative examples.
and initial negative example set N0,the SVM classifier C0 is learned, the negative example set N1 consists of the support vectors of C0 and the unlabeled examples predicted as negative examples. ∪ N1, the SVM classifier C1 is learned, the negative example set N2 consists of the support vectors of C1 and the unlabeled examples predicted as negative examples.
∪ N1, the SVM classifier C1 is learned, the negative example set N2 consists of the support vectors of C1 and the unlabeled examples predicted as negative examples.
 | is meet.
| is meet.References
-
- Lanckriet G, Deng M, Cristianini M, Jordan M, Noble W. Kernel-based data fusion and its application to protein function prediction in yeast. In Bioinformatics, Pac Symp Biocomput. 2004. pp. 300–11. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
