

Memory deficits due to familial British dementia BRI2 mutation are caused by loss of BRI2 function rather than amyloidosis
- PMID: 21048150
- PMCID: PMC3056548
- DOI: 10.1523/JNEUROSCI.3917-10.2010
Memory deficits due to familial British dementia BRI2 mutation are caused by loss of BRI2 function rather than amyloidosis
Abstract
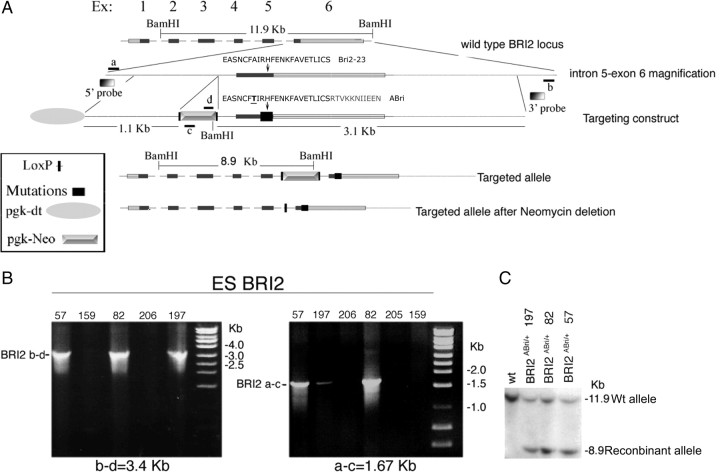
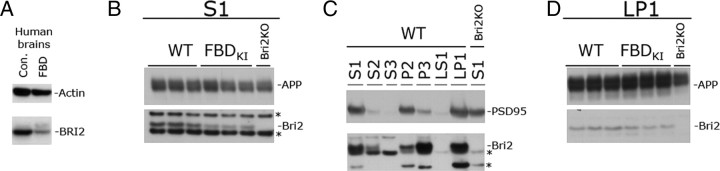
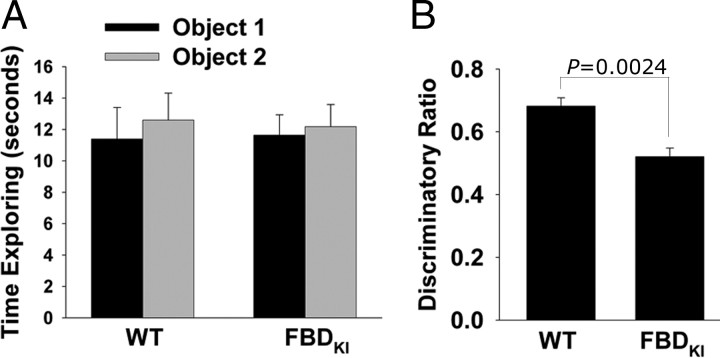
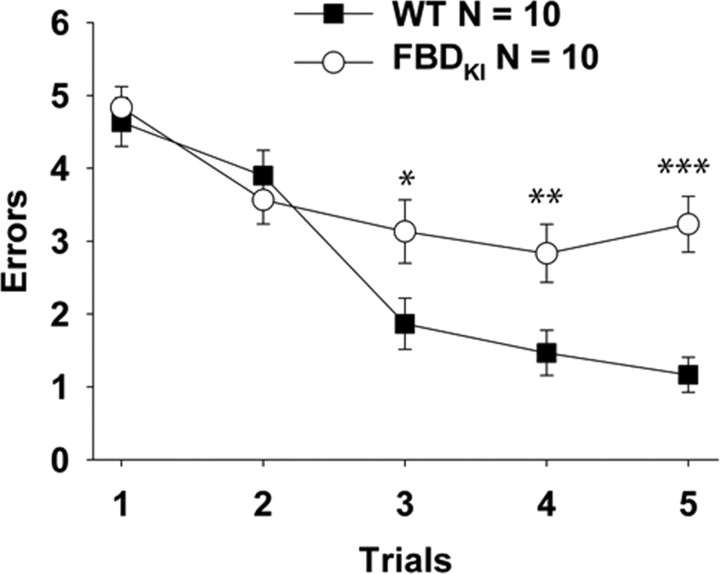
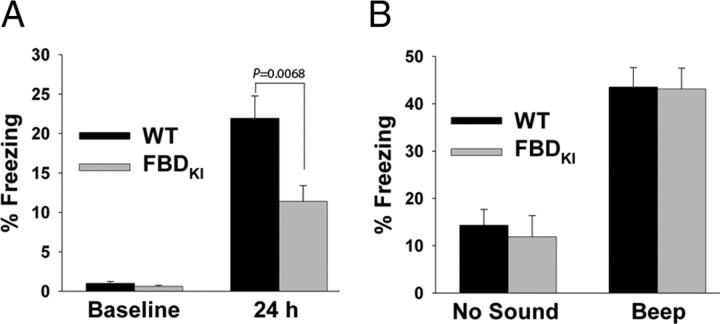
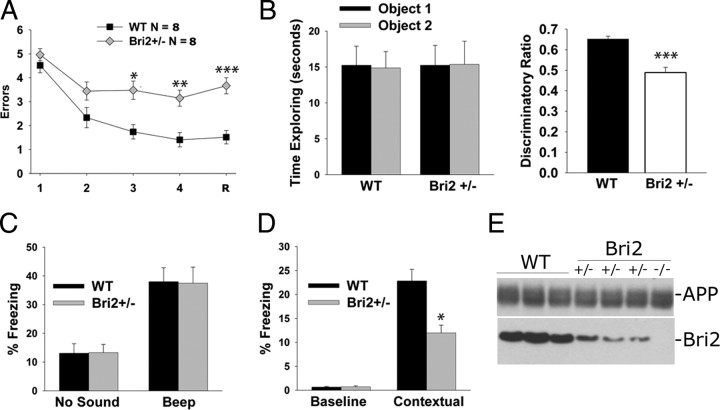
Familial dementias, which include Alzheimer disease (AD), familial British dementia (FBD), and familial Danish dementia (FDD), are caused by dominantly inherited autosomal mutations and are characterized by the production of amyloidogenic peptides, neurofibrillary tangles (NFTs) and neurodegeneration (St George-Hyslop and Petit, 2005; Garringer et al., 2009). The prevailing pathogenic theory, the "amyloid cascade hypothesis" (Hardy and Selkoe, 2002), posits that the accumulation of amyloidogenic peptides triggers tauopathy, neurodegeneration, and cognitive and behavioral changes. However, this hypothesis is yet to be validated, and causes of dementia may be multifaceted and involve other mechanisms, such as loss of function due to pathogenic mutations. Mouse models of human dementia invariably use transgenic expression systems (LaFerla and Oddo, 2005; McGowan et al., 2006; Vidal et al., 2009; Coomaraswamy et al., 2010) that do not reflect the genotypes of human disease and cannot replicate loss of function. Therefore, we generated a knock-in (KI) mouse model of FBD (FBD(KI)) genetically congruous with the human disease. FBD is caused by a missense mutation at the stop codon of the BRI2 gene (Vidal et al., 1999) and, like FBD patients, FBD(KI) mice carry this mutation in one of the two murine Bri2 alleles. We report that the British mutation drastically reduces expression of mature BRI2 in both KI mice and human FBD brains. This deficit is associated with severe hippocampal memory deficits in FBD(KI) mice. Remarkably, these animals showed no cerebral amyloidosis and tauopathy. Bri2(+/-) mice present memory deficits similar to those in FBD(KI) animals. Collectively, these results indicate that the British BRI2 mutation underlies abnormal memory due to loss of BRI2 function and independently of histopathological alterations typically evident in advanced neurodegenerative disease.
Figures
References
-
- Akiyama H, Kondo H, Arai T, Ikeda K, Kato M, Iseki E, Schwab C, McGeer PL. Expression of BRI, the normal precursor of the amyloid protein of familial British dementia, in human brain. Acta Neuropathol. 2004;107:53–58. - PubMed
-
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial nonmatching-to-sample learning task to study ‘recognition memory.’. Nat Protoc. 2006;1:1306–1311. - PubMed
-
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. - PubMed
-
- Coomaraswamy J, Kilger E, Wölfing H, Schäfer C, Kaeser SA, Wegenast-Braun BM, Hefendehl JK, Wolburg H, Mazzella M, Ghiso J, Goedert M, Akiyama H, Garcia-Sierra F, Wolfer DP, Mathews PM, Jucker M. Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:7969–7974. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases