

Microbiome profiling by illumina sequencing of combinatorial sequence-tagged PCR products
- PMID: 21048977
- PMCID: PMC2964327
- DOI: 10.1371/journal.pone.0015406
Microbiome profiling by illumina sequencing of combinatorial sequence-tagged PCR products
Abstract
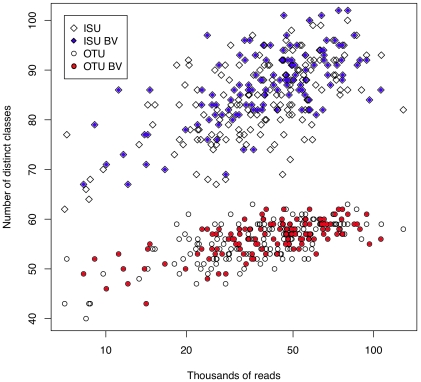
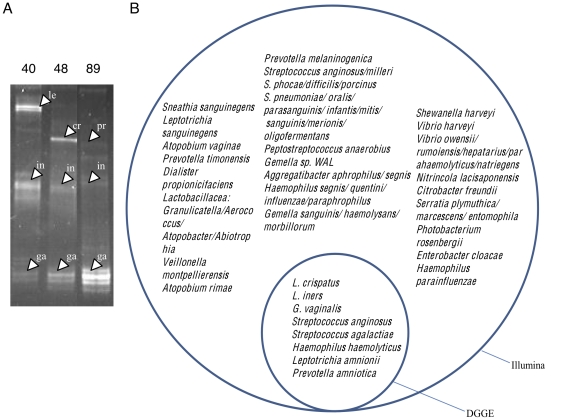
We developed a low-cost, high-throughput microbiome profiling method that uses combinatorial sequence tags attached to PCR primers that amplify the rRNA V6 region. Amplified PCR products are sequenced using an Illumina paired-end protocol to generate millions of overlapping reads. Combinatorial sequence tagging can be used to examine hundreds of samples with far fewer primers than is required when sequence tags are incorporated at only a single end. The number of reads generated permitted saturating or near-saturating analysis of samples of the vaginal microbiome. The large number of reads allowed an in-depth analysis of errors, and we found that PCR-induced errors composed the vast majority of non-organism derived species variants, an observation that has significant implications for sequence clustering of similar high-throughput data. We show that the short reads are sufficient to assign organisms to the genus or species level in most cases. We suggest that this method will be useful for the deep sequencing of any short nucleotide region that is taxonomically informative; these include the V3, V5 regions of the bacterial 16S rRNA genes and the eukaryotic V9 region that is gaining popularity for sampling protist diversity.
Conflict of interest statement
Figures


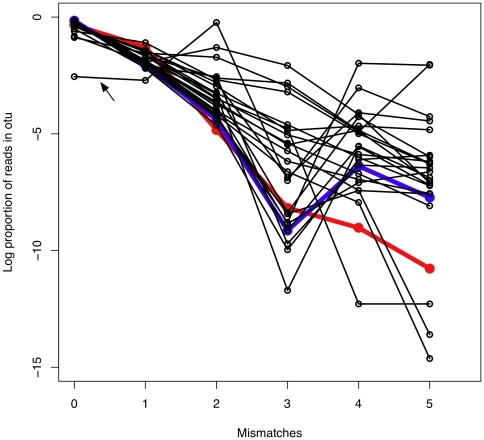


 scores a log-odds score of the likelihood of error in the base call, higher
scores a log-odds score of the likelihood of error in the base call, higher  scores represent lower likelihoods of error . They are expected to decrease with distance from the left or right sequencing primer, and to be highest in the region of perfect overlap because
scores represent lower likelihoods of error . They are expected to decrease with distance from the left or right sequencing primer, and to be highest in the region of perfect overlap because  scores are additive.
scores are additive.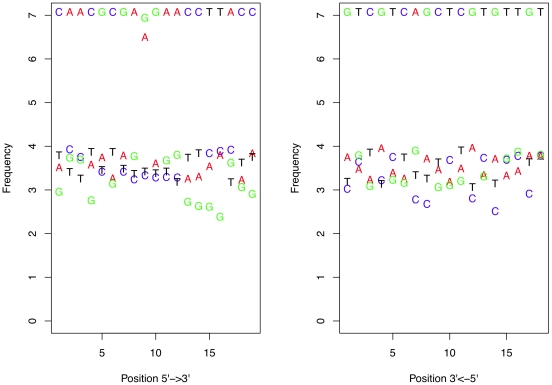
 million sequences, and the difference in frequency between the correct and altered nucleotide is relatively constant. Note that the errors are at the same frequency at each end of the primers.
million sequences, and the difference in frequency between the correct and altered nucleotide is relatively constant. Note that the errors are at the same frequency at each end of the primers.

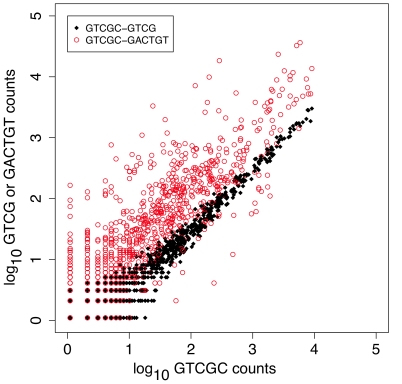

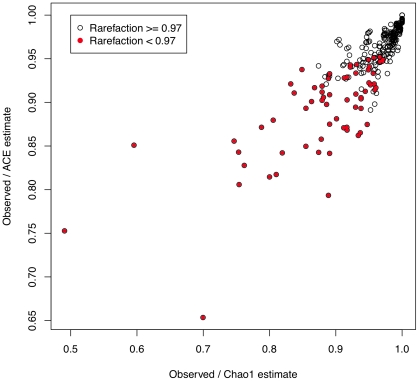
References
-
- Polymenakou PN, Lampadariou N, Mandalakis M, Tselepides A. Phylogenetic diversity of sediment bacteria from the southern cretan margin, eastern mediterranean sea. Syst Appl Microbiol. 2009 Feb;32:17–26. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
