

Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman's syndrome
- PMID: 21051746
- PMCID: PMC3082423
- DOI: 10.2215/CJN.06730810
Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman's syndrome
Abstract
Background and objectives: Gitelman's syndrome (GS) is an autosomal recessive renal tubular disorder caused by mutations in the SLC12A3 gene encoding the thiazide-sensitive Na(+)-Cl(-) cotransporter (NCC). Despite meticulous sequencing of genomic DNA, approximately one-third of GS patients are negative or heterozygotes for the known mutations.
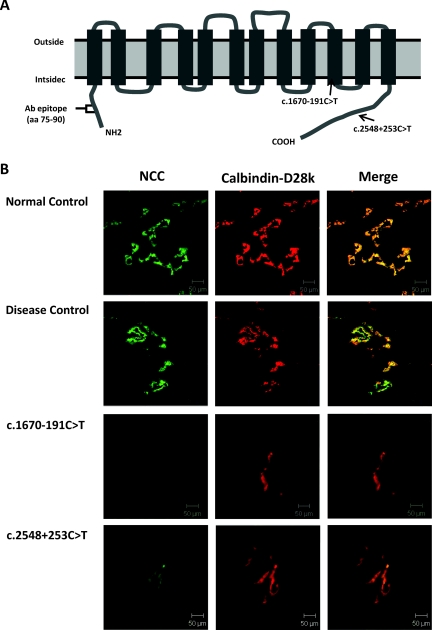
Design, setting, participants, & measurements: Because blood leukocytes express NCC mRNA, we evaluate whether deep intronic mutations contribute to GS patients with uniallelic or undetectable SLC12A3 mutations. Twenty-nine patients with GS (men/women = 16/13), including eight negative and 21 uniallelic SLC12A3 mutations from 19 unrelated families, and normal controls were enrolled in an academic medical center. Analysis of cDNA from blood leukocytes, sequencing of the corresponding introns of genomic DNA for abnormal transcript, and analysis of NCC protein expression from renal biopsy were performed.
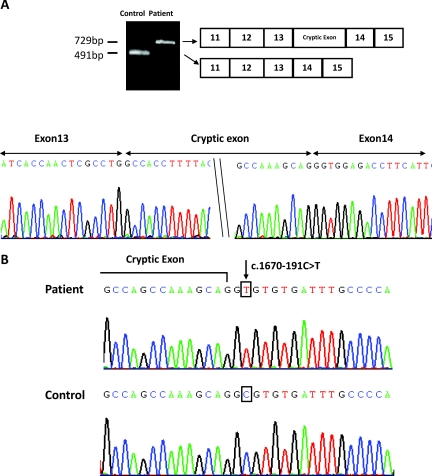
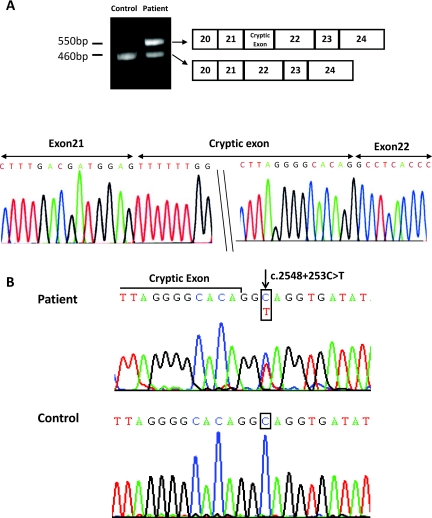
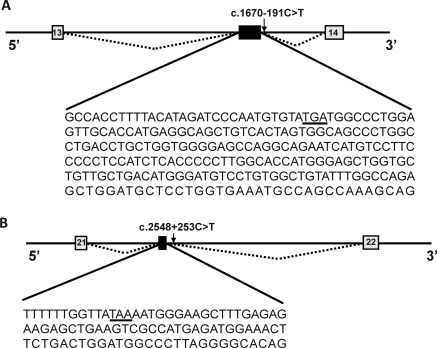
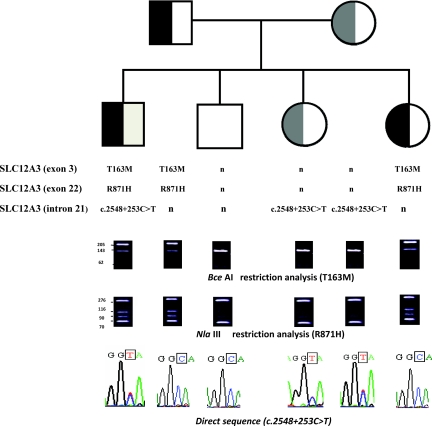
Results: We identified nine Taiwan aboriginal patients carrying c.1670-191C→T mutations in intron 13 and 10 nonaboriginal patients carrying c.2548+253C→T mutations in intron 21 from 14 families (14/19). These two mutations undetected in 100 healthy subjects created pseudoexons containing new premature termination codons. Haplotype analysis with markers flanking SLC12A3 revealed that both mutations did not have founder effects. Apical NCC expression in the DCT of renal tissue was markedly diminished in two patients carrying deep intronic mutations.
Conclusions: Deep intronic mutations in SLC12A3 causing defective NCC expression can be identified with the RNA-based approach in patients with GS. c.1670-191C→T and c.2548+253C→T are hot spot mutations that can be screened in GS patients with uniallelic or negative SLC12A3 mutations.
Figures
References
-
- Gitelman HJ, Graham JB, Welt GL: A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 79: 211–235, 1996 - PubMed
-
- Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB: Gitelman's syndrome revisited: An evaluation of symptoms and health-related quality of life. Kidney Int 59: 710–717, 2001 - PubMed
-
- Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP: Gitelman's variant of Bartter's syndrome, inherited hypokalemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996 - PubMed
-
- Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, Jonh E, Lifton RP: Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat Genet 17: 171–178, 1997 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
