

The autophagic tumor stroma model of cancer or "battery-operated tumor growth": A simple solution to the autophagy paradox
- PMID: 21051947
- PMCID: PMC3055183
- DOI: 10.4161/cc.9.21.13817
The autophagic tumor stroma model of cancer or "battery-operated tumor growth": A simple solution to the autophagy paradox
Abstract
The role of autophagy in tumorigenesis is controversial. Both autophagy inhibitors (chloroquine) and autophagy promoters (rapamycin) block tumorigenesis by unknown mechanism(s). This is called the "Autophagy Paradox". We have recently reported a simple solution to this paradox. We demonstrated that epithelial cancer cells use oxidative stress to induce autophagy in the tumor microenvironment. As a consequence, the autophagic tumor stroma generates recycled nutrients that can then be used as chemical building blocks by anabolic epithelial cancer cells. This model results in a net energy transfer from the tumor stroma to epithelial cancer cells (an energy imbalance), thereby promoting tumor growth. This net energy transfer is both unilateral and vectorial, from the tumor stroma to the epithelial cancer cells, representing a true host-parasite relationship. We have termed this new paradigm "The Autophagic Tumor Stroma Model of Cancer Cell Metabolism" or "Battery-Operated Tumor Growth". In this sense, autophagy in the tumor stroma serves as a "battery" to fuel tumor growth, progression and metastasis, independently of angiogenesis. Using this model, the systemic induction of autophagy will prevent epithelial cancer cells from using recycled nutrients, while the systemic inhibiton of autophagy will prevent stromal cells from producing recycled nutrients-both effectively "starving" cancer cells. We discuss the idea that tumor cells could become resistant to the systemic induction of autophagy, by the upregulation of natural endogenous autophagy inhibitors in cancer cells. Alternatively, tumor cells could also become resistant to the systemic induction of autophagy, by the genetic silencing/deletion of pro-autophagic molecules, such as Beclin1. If autophagy resistance develops in cancer cells, then the systemic inhibition of autophagy would provide a therapeutic solution to this type of drug resistance, as it would still target autophagy in the tumor stroma. As such, an anti-cancer therapy that combines the alternating use of both autophagy promoters and autophagy inhibitors would be expected to prevent the onset of drug resistance. We also discuss why anti-angiogenic therapy has been found to promote tumor recurrence, progression and metastasis. More specifically, anti-angiogenic therapy would induce autophagy in the tumor stroma via the induction of stromal hypoxia, thereby converting a non-aggressive tumor type to a "lethal" aggressive tumor phenotype. Thus, uncoupling the metabolic parasitic relationship between cancer cells and an autophagic tumor stroma may hold great promise for anti-cancer therapy. Finally, we believe that autophagy in the tumor stroma is the local microscopic counterpart of systemic wasting (cancer-associated cachexia), which is associated with advanced and metastatic cancers. Cachexia in cancer patients is not due to decreased energy intake, but instead involves an increased basal metabolic rate and increased energy expenditures, resulting in a negative energy balance. Importantly, when tumors were surgically excised, this increased metabolic rate returned to normal levels. This view of cachexia, resulting in energy transfer to the tumor, is consistent with our hypothesis. So, cancer-associated cachexia may start locally as stromal autophagy, and then spread systemically. As such, stromal autophagy may be the requisite precursor of systemic cancer-associated cachexia.
Figures
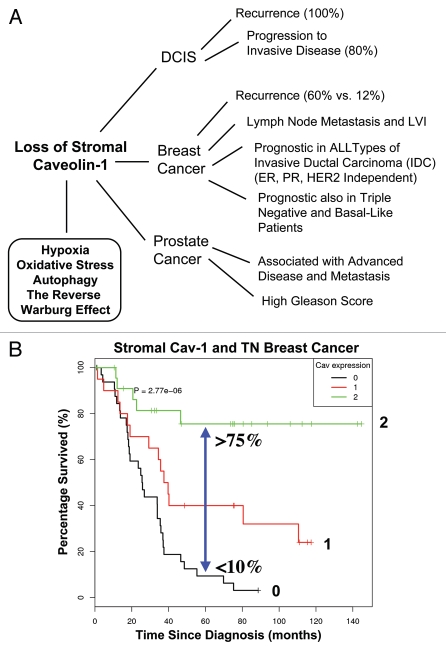





References
-
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–3276. - PMC - PubMed
-
- Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–3551. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA075503/CA/NCI NIH HHS/United States
- R01 CA098779/CA/NCI NIH HHS/United States
- R01-CA-120876/CA/NCI NIH HHS/United States
- R01 CA120876/CA/NCI NIH HHS/United States
- R01-CA-70896/CA/NCI NIH HHS/United States
- R01-CA-098779/CA/NCI NIH HHS/United States
- R01-CA-86072/CA/NCI NIH HHS/United States
- R01-AR-055660/AR/NIAMS NIH HHS/United States
- R01-CA-080250/CA/NCI NIH HHS/United States
- R01 CA070896/CA/NCI NIH HHS/United States
- R01 CA107382/CA/NCI NIH HHS/United States
- P30 CA056036/CA/NCI NIH HHS/United States
- P30-CA-56036/CA/NCI NIH HHS/United States
- R01-CA-107382/CA/NCI NIH HHS/United States
- R01 AR055660/AR/NIAMS NIH HHS/United States
- R01-CA-75503/CA/NCI NIH HHS/United States
- R01 CA080250/CA/NCI NIH HHS/United States
- R01 CA086072/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials