

Molecular characterization of apoptosis induced by CARF silencing in human cancer cells
- PMID: 21052095
- PMCID: PMC3131902
- DOI: 10.1038/cdd.2010.129
Molecular characterization of apoptosis induced by CARF silencing in human cancer cells
Erratum in
- Cell Death Differ. 2011 Jul;18(7):1238
Abstract
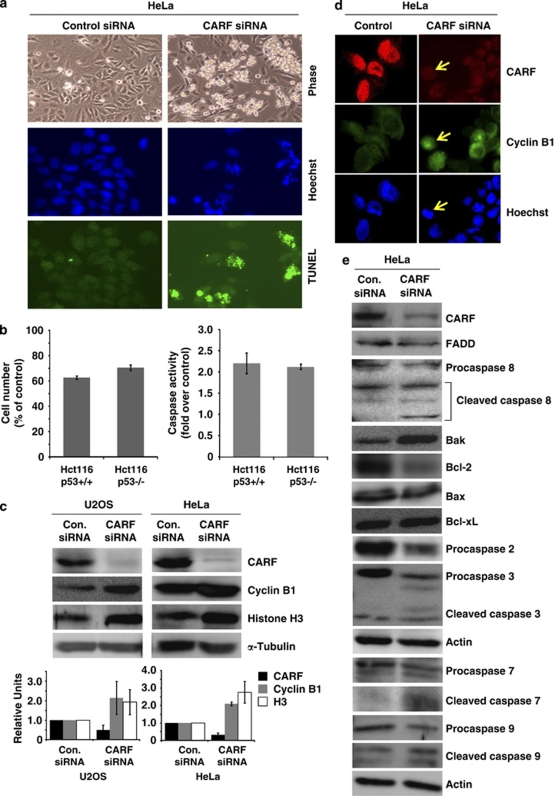
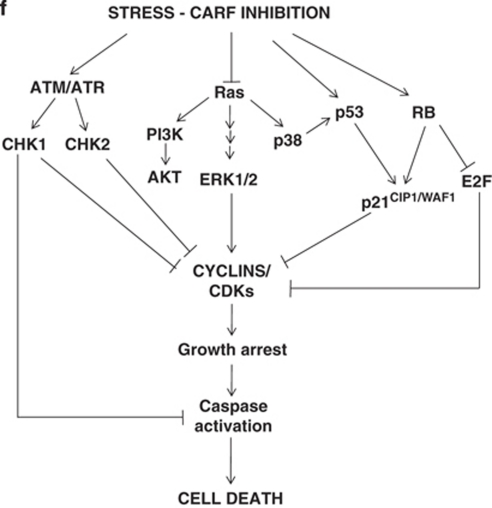
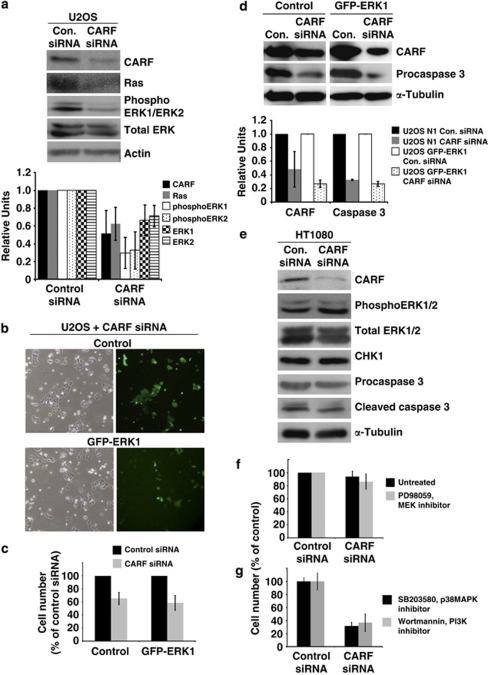
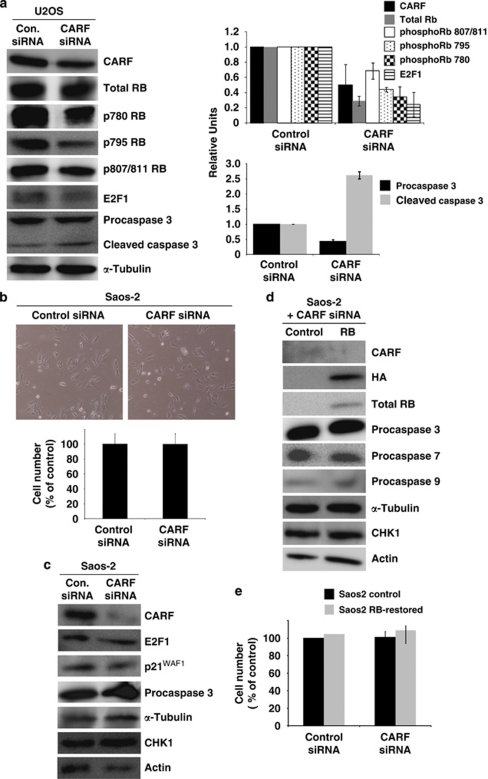
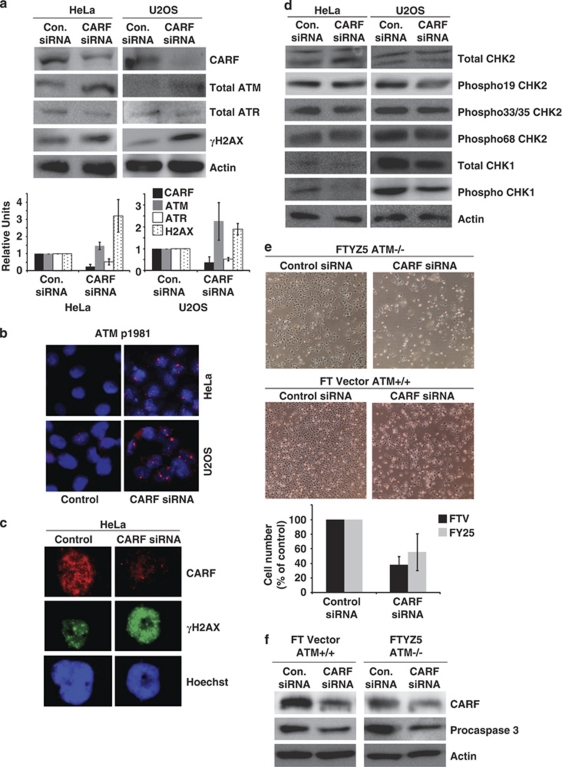
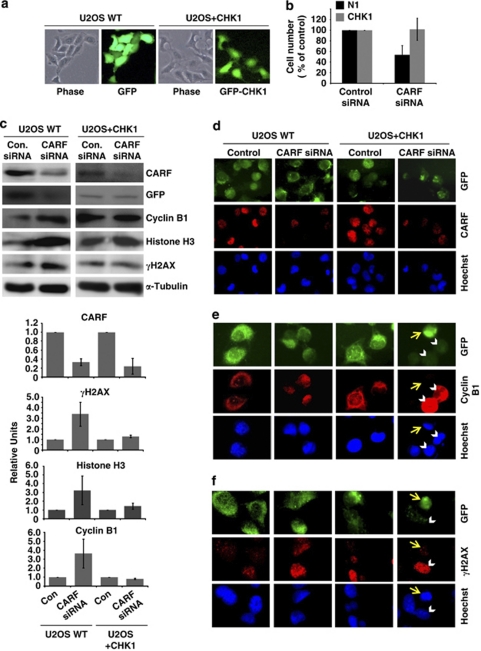
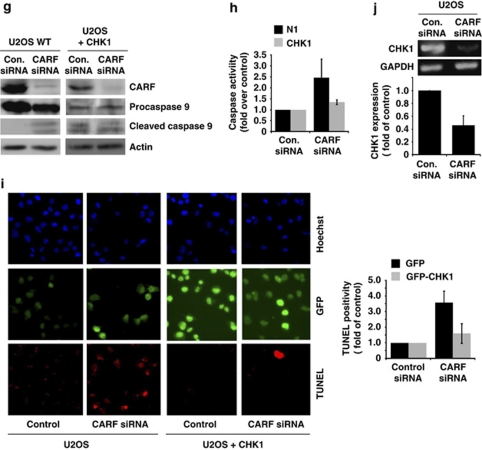
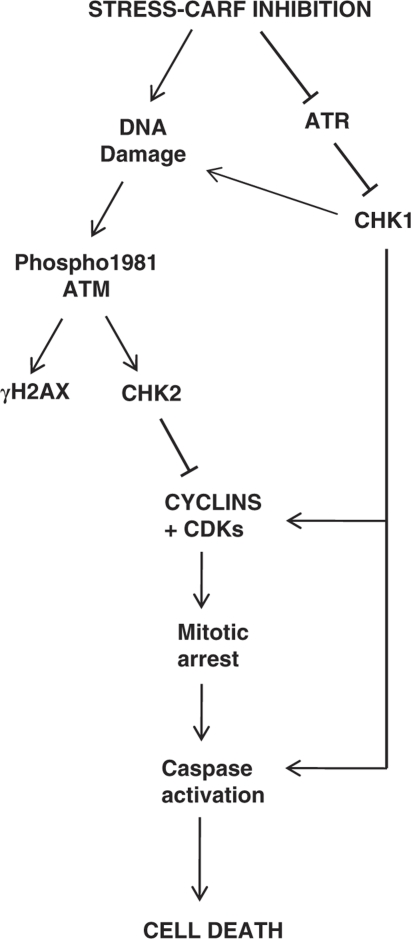
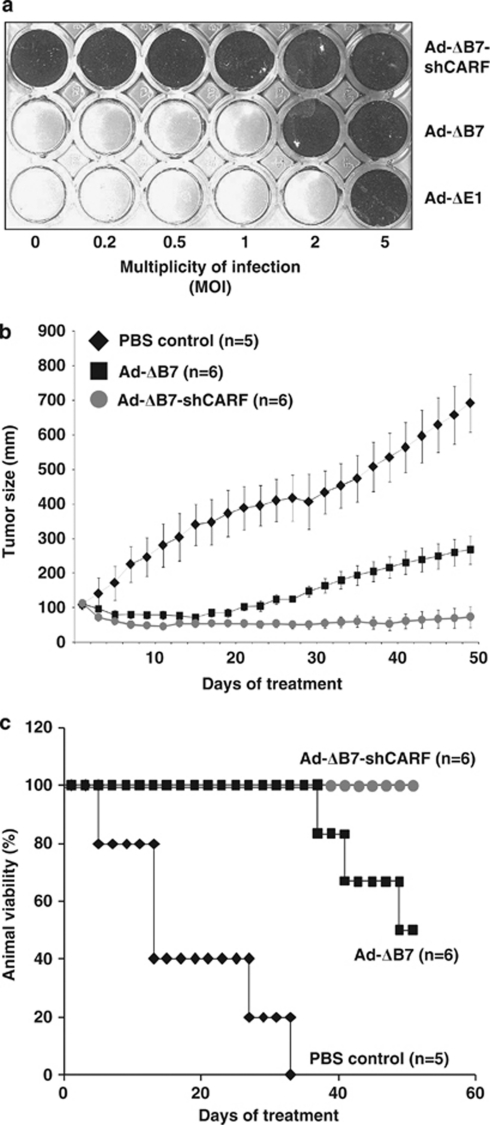
Collaborator of ARF (CARF) was cloned as an ARF-interacting protein and shown to regulate the p53-p21(WAF1)-HDM2 pathway, which is central to tumor suppression via senescence and apoptosis. We had previously reported that CARF inhibition in cancer cells led to polyploidy and caspase-dependent apoptosis, however, the mechanisms governing this phenomenon remained unknown. Thus, we examined various cell death and survival pathways including the mitochondrial stress, ataxia telangiectasia mutated (ATM)-ATR, Ras-MAP kinase and retinoblastoma cascades. We found that CARF is a pleiotropic regulator with widespread effects; its suppression affected all investigated pathways. Most remarkably, it protected the cells against genotoxicity; CARF knockdown elicited DNA damage response as evidenced by increased levels of phosphorylated ATM and γH2AX, leading to induction of mitotic arrest and eventual apoptosis. We also show that the CARF-silencing-induced apoptosis in vitro translates to in vivo. In a human tumor xenograft mouse model, treatment of developing tumors with short hairpin RNA (shRNA) against CARF via an adenovirus carrier induced complete suppression of tumor growth, suggesting that CARF shRNA is a strong candidate for an anticancer reagent. We demonstrate that CARF has a vital role in genome preservation and tumor suppression and CARF siRNA is an effective novel cancer therapeutic agent.
© 2011 Macmillan Publishers Limited
Figures
Similar articles
-
Collaborator of ARF (CARF) regulates proliferative fate of human cells by dose-dependent regulation of DNA damage signaling.J Biol Chem. 2014 Jun 27;289(26):18258-69. doi: 10.1074/jbc.M114.547208. Epub 2014 May 13. J Biol Chem. 2014. PMID: 24825908 Free PMC article.
-
Molecular characterization of collaborator of ARF (CARF) as a DNA damage response and cell cycle checkpoint regulatory protein.Exp Cell Res. 2014 Apr 1;322(2):324-34. doi: 10.1016/j.yexcr.2014.01.022. Epub 2014 Jan 28. Exp Cell Res. 2014. PMID: 24485912
-
CARF (Collaborator of ARF) overexpression in p53-deficient cells promotes carcinogenesis.Mol Oncol. 2015 Nov;9(9):1877-89. doi: 10.1016/j.molonc.2015.07.003. Epub 2015 Aug 4. Mol Oncol. 2015. PMID: 26278998 Free PMC article.
-
CARF is a multi-module regulator of cell proliferation and a molecular bridge between cellular senescence and carcinogenesis.Mech Ageing Dev. 2017 Sep;166:64-68. doi: 10.1016/j.mad.2017.07.008. Epub 2017 Jul 25. Mech Ageing Dev. 2017. PMID: 28754531 Review.
-
Molecular bridging of aging and cancer: A CARF link.Ann N Y Acad Sci. 2010 Jun;1197:129-33. doi: 10.1111/j.1749-6632.2009.05392.x. Ann N Y Acad Sci. 2010. PMID: 20536841 Review.
Cited by
-
Tumor suppressor activity of miR-451: Identification of CARF as a new target.Sci Rep. 2018 Jan 10;8(1):375. doi: 10.1038/s41598-017-18559-5. Sci Rep. 2018. PMID: 29321561 Free PMC article.
-
NR4A3 inhibits the tumor progression of hepatocellular carcinoma by inducing cell cycle G0/G1 phase arrest and upregulation of CDKN2AIP expression.Int J Biol Sci. 2024 Oct 28;20(15):5850-5867. doi: 10.7150/ijbs.95174. eCollection 2024. Int J Biol Sci. 2024. PMID: 39664575 Free PMC article.
-
Soyasapogenol-A targets CARF and results in suppression of tumor growth and metastasis in p53 compromised cancer cells.Sci Rep. 2020 Apr 14;10(1):6323. doi: 10.1038/s41598-020-62953-5. Sci Rep. 2020. PMID: 32286347 Free PMC article.
-
Chemical biology reveals CARF as a positive regulator of canonical Wnt signaling by promoting TCF/β-catenin transcriptional activity.Cell Discov. 2017 Jan 31;3:17003. doi: 10.1038/celldisc.2017.3. eCollection 2017. Cell Discov. 2017. PMID: 28417011 Free PMC article.
-
Differential activities of the two closely related withanolides, Withaferin A and Withanone: bioinformatics and experimental evidences.PLoS One. 2012;7(9):e44419. doi: 10.1371/journal.pone.0044419. Epub 2012 Sep 4. PLoS One. 2012. PMID: 22973447 Free PMC article.
References
-
- Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. - PubMed
-
- Hasan MK, Yaguchi T, Sugihara T, Kumar PK, Taira K, Reddel RR, et al. CARF is a novel protein that cooperates with mouse p19ARF (human p14ARF) in activating p53. J Biol Chem. 2002;277:37765–37770. - PubMed
-
- Hasan K, Cheung C, Kaul Z, Shah N, Sakaushi S, Sugimoto K, et al. CARF is a vital dual regulator of cellular senescence and apoptosis. J Biol Chem. 2009;284:1664–1672. - PubMed
-
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: A molecular definition. Oncogene. 2004;23:2825–2837. - PubMed
-
- Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
