

Genome-wide fitness and genetic interactions determined by Tn-seq, a high-throughput massively parallel sequencing method for microorganisms
- PMID: 21053251
- PMCID: PMC3877651
- DOI: 10.1002/9780471729259.mc01e03s19
Genome-wide fitness and genetic interactions determined by Tn-seq, a high-throughput massively parallel sequencing method for microorganisms
Abstract
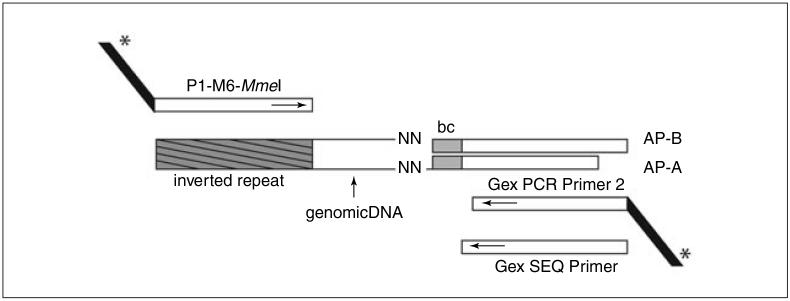
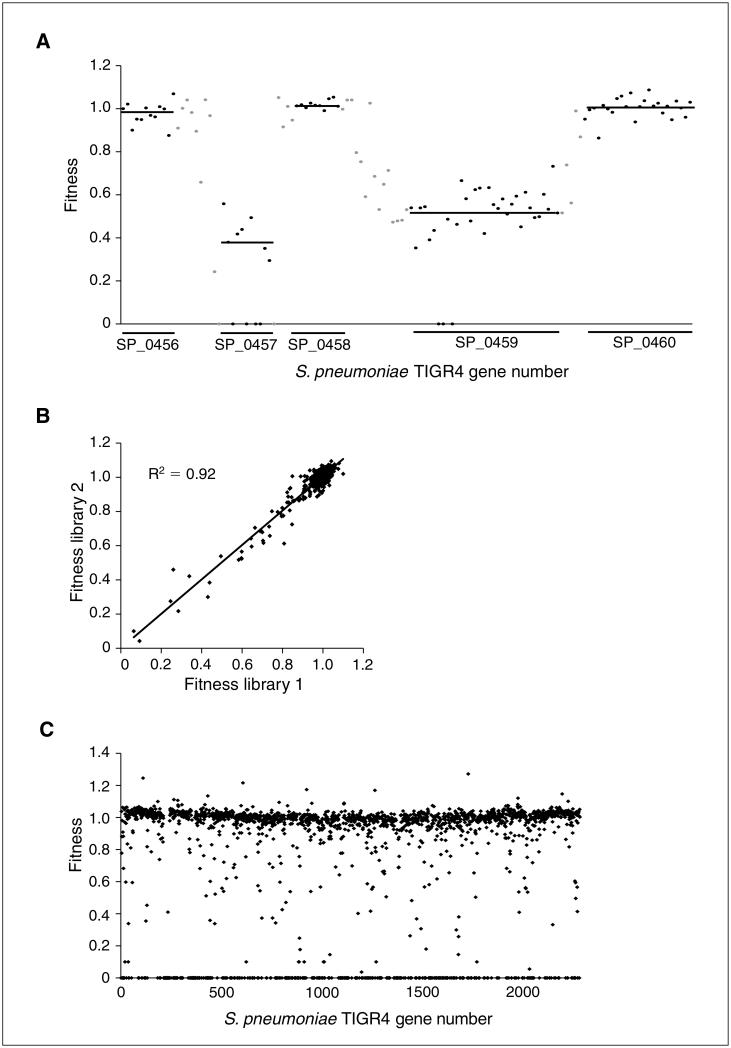
The lagging annotation of bacterial genomes and the inherent genetic complexity of many phenotypes is hindering the discovery of new drug targets and the development of new antimicrobials and vaccines. Here we present the method Tn-seq, with which it has become possible to quantitatively determine fitness for most genes in a microorganism and to screen for quantitative genetic interactions on a genome-wide scale and in a high-throughput fashion. Tn-seq can thus direct studies in the annotation of genes and untangle complex phenotypes. The method is based on the construction of a saturated Mariner transposon insertion library. After library selection, changes in frequency of each insertion mutant are determined by sequencing of the flanking regions en masse. These changes are used to calculate each mutant's fitness. The method has been developed for the Gram-positive bacterium Streptococcus pneumoniae, a causative agent of pneumonia and meningitis; however, due to the wide activity of the Mariner transposon, Tn-seq can be applied to many different microbial species.
Figures

Similar articles
-
Genome-Wide Fitness and Genetic Interactions Determined by Tn-seq, a High-Throughput Massively Parallel Sequencing Method for Microorganisms.Curr Protoc Microbiol. 2015 Feb 2;36:1E.3.1-1E.3.24. doi: 10.1002/9780471729259.mc01e03s36. Curr Protoc Microbiol. 2015. PMID: 25641100 Free PMC article.
-
Genome-Wide Fitness and Genetic Interactions Determined by Tn-seq, a High-Throughput Massively Parallel Sequencing Method for Microorganisms.Curr Protoc Mol Biol. 2014 Apr 14;106:7.16.1-7.16.24. doi: 10.1002/0471142727.mb0716s106. Curr Protoc Mol Biol. 2014. PMID: 24733243 Free PMC article. Review.
-
Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms.Nat Methods. 2009 Oct;6(10):767-72. doi: 10.1038/nmeth.1377. Epub 2009 Sep 20. Nat Methods. 2009. PMID: 19767758 Free PMC article.
-
High-Throughput Mutant Screening via Transposon Sequencing.Cold Spring Harb Protoc. 2023 Oct 3;2023(10):707-9. doi: 10.1101/pdb.top107867. Cold Spring Harb Protoc. 2023. PMID: 36931734 Free PMC article.
-
Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms.Nat Rev Microbiol. 2013 Jul;11(7):435-42. doi: 10.1038/nrmicro3033. Epub 2013 May 28. Nat Rev Microbiol. 2013. PMID: 23712350 Free PMC article. Review.
Cited by
-
Temporal genome-wide fitness analysis of Mycobacterium marinum during infection reveals the genetic requirement for virulence and survival in amoebae and microglial cells.mSystems. 2024 Feb 20;9(2):e0132623. doi: 10.1128/msystems.01326-23. Epub 2024 Jan 25. mSystems. 2024. PMID: 38270456 Free PMC article.
-
Tn-seq explorer: a tool for analysis of high-throughput sequencing data of transposon mutant libraries.PLoS One. 2015 May 4;10(5):e0126070. doi: 10.1371/journal.pone.0126070. eCollection 2015. PLoS One. 2015. PMID: 25938432 Free PMC article.
-
Integrated genomics and chemical biology herald an era of sophisticated antibacterial discovery, from defining essential genes to target elucidation.Cell Chem Biol. 2022 May 19;29(5):716-729. doi: 10.1016/j.chembiol.2022.04.006. Epub 2022 May 5. Cell Chem Biol. 2022. PMID: 35523184 Free PMC article. Review.
-
Bacterial Factors Required for Transmission of Streptococcus pneumoniae in Mammalian Hosts.Cell Host Microbe. 2019 Jun 12;25(6):884-891.e6. doi: 10.1016/j.chom.2019.04.012. Epub 2019 May 21. Cell Host Microbe. 2019. PMID: 31126758 Free PMC article.
-
Genomewide Identification of Essential Genes and Fitness Determinants of Streptococcus mutans UA159.mSphere. 2018 Feb 7;3(1):e00031-18. doi: 10.1128/mSphere.00031-18. eCollection 2018 Jan-Feb. mSphere. 2018. PMID: 29435491 Free PMC article.
References
-
- Bae T, Glass EM, Schneewind O, Missiakas D. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Molec. Biol. 2008;416:103–116. - PubMed
-
- Begley TJ, Rosenbach AS, Ideker T, Samson LD. Damage recovery pathways in Saccharomyces cerevisiae revealed by genomic phenotyping and interactome mapping. Molec. Cancer Res. 2002;1:103–112. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
