

Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females
- PMID: 21053371
- PMCID: PMC3033517
- DOI: 10.1002/humu.21373
Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females
Abstract
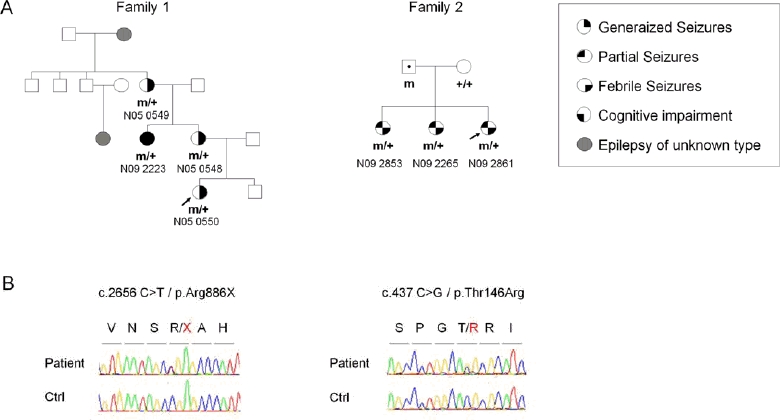
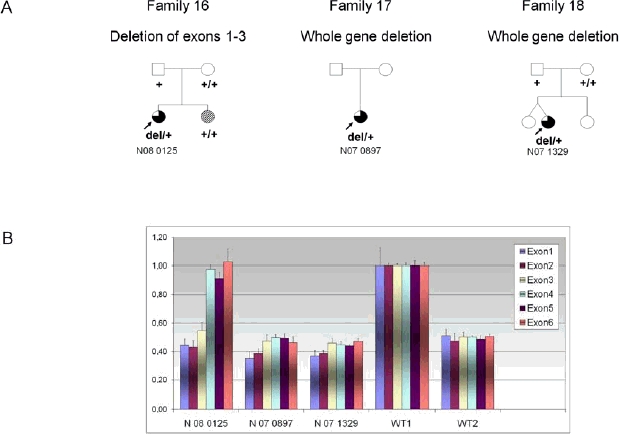
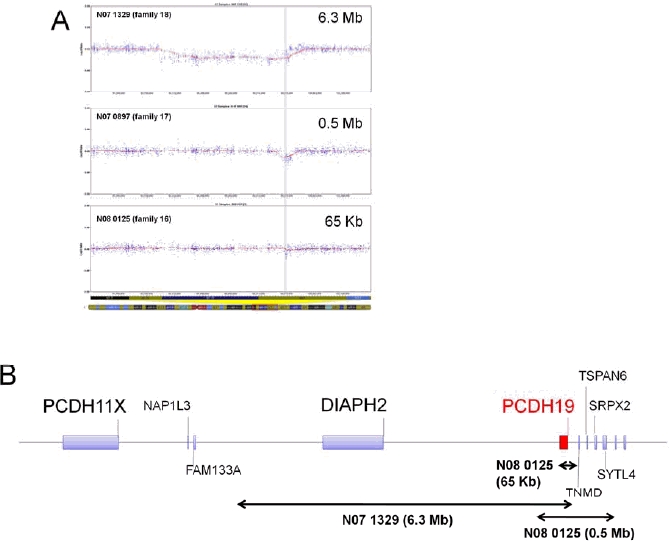
Mutations in PCDH19, encoding protocadherin 19 on chromosome X, cause familial epilepsy and mental retardation limited to females or Dravet-like syndrome. Heterozygous females are affected while hemizygous males are spared, this unusual mode of inheritance being probably due to a mechanism called cellular interference. To extend the mutational and clinical spectra associated with PCDH19, we screened 150 unrelated patients (113 females) with febrile and afebrile seizures for mutations or rearrangements in the gene. Fifteen novel point mutations were identified in 15 female patients (6 sporadic and 9 familial cases). In addition, qPCR revealed two whole gene deletions and one partial deletion in 3 sporadic female patients. Clinical features were highly variable but included almost constantly a high sensitivity to fever and clusters of brief seizures. Interestingly, cognitive functions were normal in several family members of 2 families: the familial condition in family 1 was suggestive of Generalized Epilepsy with Febrile Seizures Plus (GEFS+) whereas all three affected females had partial cryptogenic epilepsy. These results show that mutations in PCDH19 are a relatively frequent cause of epilepsy in females and should be considered even in absence of family history and/or mental retardation.
© 2010 Wiley-Liss, Inc.
Figures
References
-
- Bione S, Sala C, Manzini C, Arrigo G, Zuffardi O, Banfi S, Borsani G, Jonveaux P, Philippe C, Zuccotti M. A human homologue of the Drosophila melanogaster diaphanous gene is disrupted in a patient with premature ovarian failure: evidence for conserved function in oogenesis and implications for human sterility. Am J Hum Genet. 1998;62:533–41. - PMC - PubMed
-
- Compagni A, Logan M, Klein R, Adams RH. Control of skeletal patterning by ephrinB1-EphB interactions. Dev Cell. 2003;5:217–30. - PubMed
-
- Depienne C, Trouillard O, Gourfinkel-An I, Saint-Martin C, Bouteiller D, Graber D, Barthez-Carpentier MA, Gautier A, Villeneuve N, Dravet C. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet. 2010;47:404–10. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
