

The DNA methylome
- PMID: 21056564
- PMCID: PMC3129437
- DOI: 10.1016/j.febslet.2010.10.061
The DNA methylome
Abstract
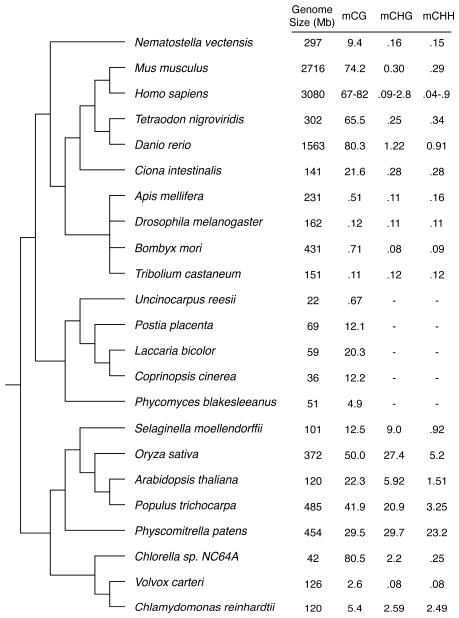
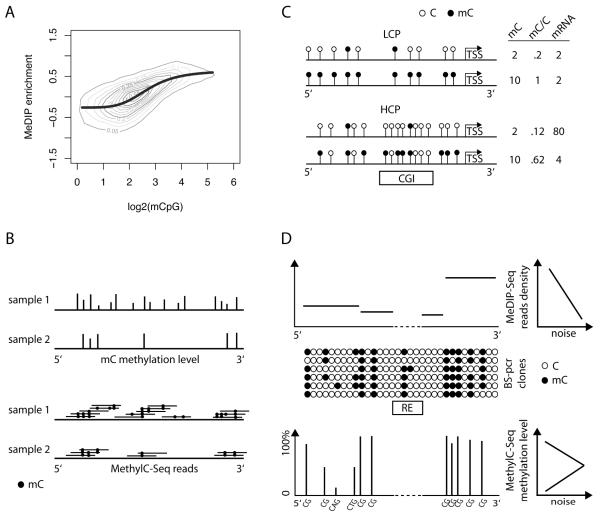
Methylation of cytosines is a pervasive feature of eukaryotic genomes and an important epigenetic layer that is fundamental for cellular differentiation processes and control of transcriptional potential. DNA methylation patterns can be inherited and influenced by the environment, diet and aging, and disrupted in diseases. Complete DNA methylomes for several organisms are now available, helping clarify the evolutionary story of this epigenetic mark and its distribution in key genomic elements. Nonetheless, a complete understanding of its role, the mechanisms responsible for its establishment and maintenance, and its cross talk with other components of cellular machinery remains elusive.
Copyright © 2010 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Holliday R, Pugh JE. DNA modification mechanis during development. Science. 1975;187:226–232. - PubMed
-
- Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. - PubMed
-
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature Reviews Genetics. 2009;10:295–304. - PubMed
-
- Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park IH, Yu J, Daley GQ, Eggan K, Hochedlinger K, Thomson J, Wang W, Gao Y, Zhang K. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. - PMC - PubMed
-
- Doi A, Park I, Wen B, Murakami P, Aryee M, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley G, Feinberg A. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–1353. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
