

Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice
- PMID: 21057039
- PMCID: PMC3023256
- DOI: 10.1152/ajpheart.00868.2010
Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice
Abstract
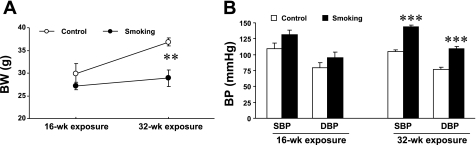
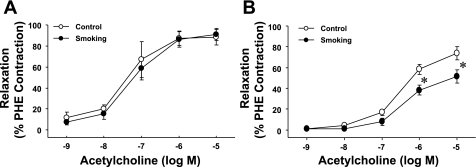
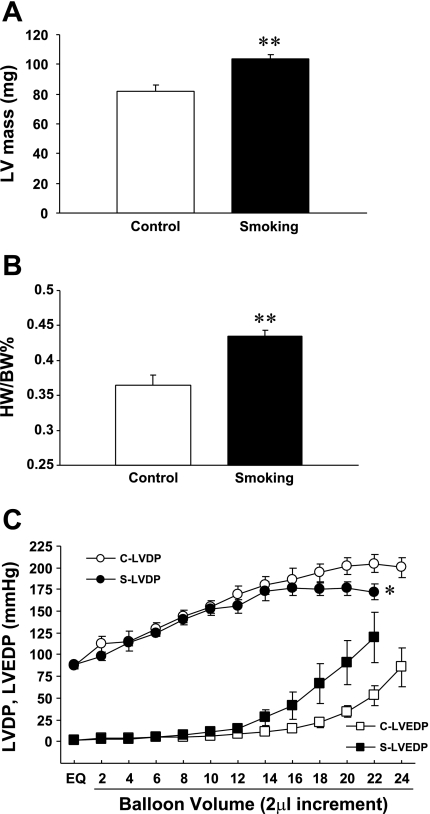
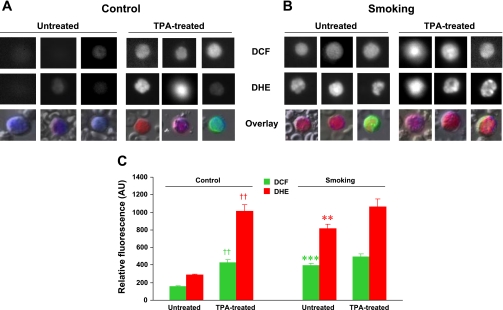
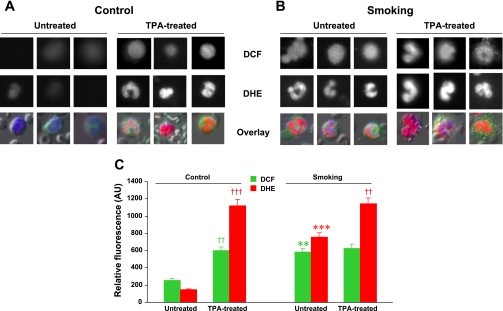
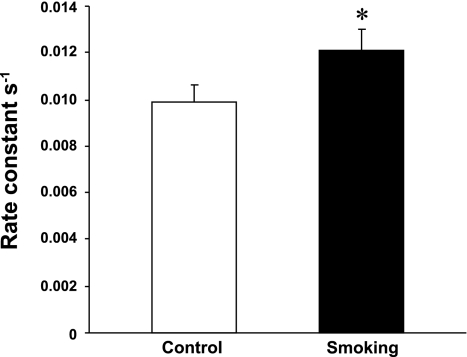
Cigarette smoking is a major independent risk factor for cardiovascular disease. While the association between chronic smoking and cardiovascular disease is well established, the underlying mechanisms are incompletely understood, partly due to the lack of adequate in vivo animal models. Here, we report a mouse model of chronic smoking-induced cardiovascular pathology. Male C57BL/6J mice were exposed to whole body mainstream cigarette smoke (CS) using a SCIREQ "InExpose" smoking system (48 min/day, 5 days/wk) for 16 or 32 wk. Age-matched, air-exposed mice served as nonsmoking controls. Blood pressure was measured, and cardiac MRI was performed. In vitro vascular ring and isolated heart experiments were performed to measure vascular reactivity and cardiac function. Blood from control and smoking mice was studied for the nitric oxide (NO) decay rate and reactive oxygen species (ROS) generation. With 32 wk of CS exposure, mice had significantly less body weight gain and markedly higher blood pressure. At 32 wk of CS exposure, ACh-induced vasorelaxation was significantly shifted to the right and downward, left ventricular mass was significantly larger along with an increased heart-to-body weight ratio, in vitro cardiac function tended to be impaired with high afterload, white blood cells had significantly higher ROS generation, and the blood NO decay rate was significantly faster. Thus, smoking led to blunted weight gain, hypertension, endothelial dysfunction, leukocyte activation with ROS generation, decreased NO bioavailability, and mild cardiac hypertrophy in mice that were not otherwise predisposed to disease. This mouse model is a useful tool to enable further elucidation of the molecular and cellular mechanisms of smoking-induced cardiovascular diseases.
Figures
References
-
- Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol 43: 1731–1737, 2004 - PubMed
-
- Barua RS, Ambrose JA, Eales-Reynolds LJ, DeVoe MC, Zervas JG, Saha DC. Dysfunctional endothelial nitric oxide biosynthesis in healthy smokers with impaired endothelium-dependent vasodilatation. Circulation 104: 1905–1910, 2001 - PubMed
-
- Bergmann S, Siekmeier R, Mix C, Jaross W. Even moderate cigarette smoking influences the pattern of circulating monocytes and the concentration of sICAM-1. Respir Physiol 114: 269–275, 1998 - PubMed
-
- Bernhard D, Wang XL. Smoking, oxidative stress and cardiovascular diseases–do anti-oxidative therapies fail? Curr Med Chem 14: 1703–1712, 2007 - PubMed
-
- Brook RD, Rajagopalan S. Particulate matter, air pollution, and blood pressure. J Am Soc Hypertens 3: 332–350, 2009 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical