

FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing
- PMID: 21057495
- PMCID: PMC3247016
- DOI: 10.1038/nmeth.1529
FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing
Abstract
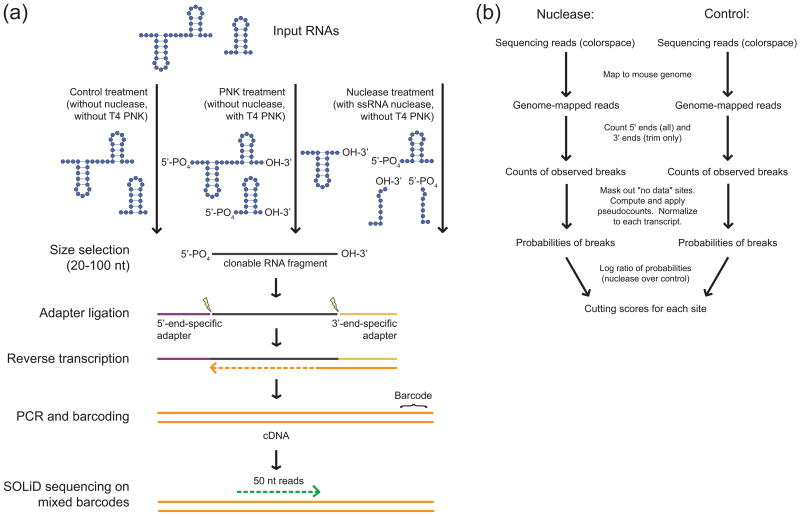
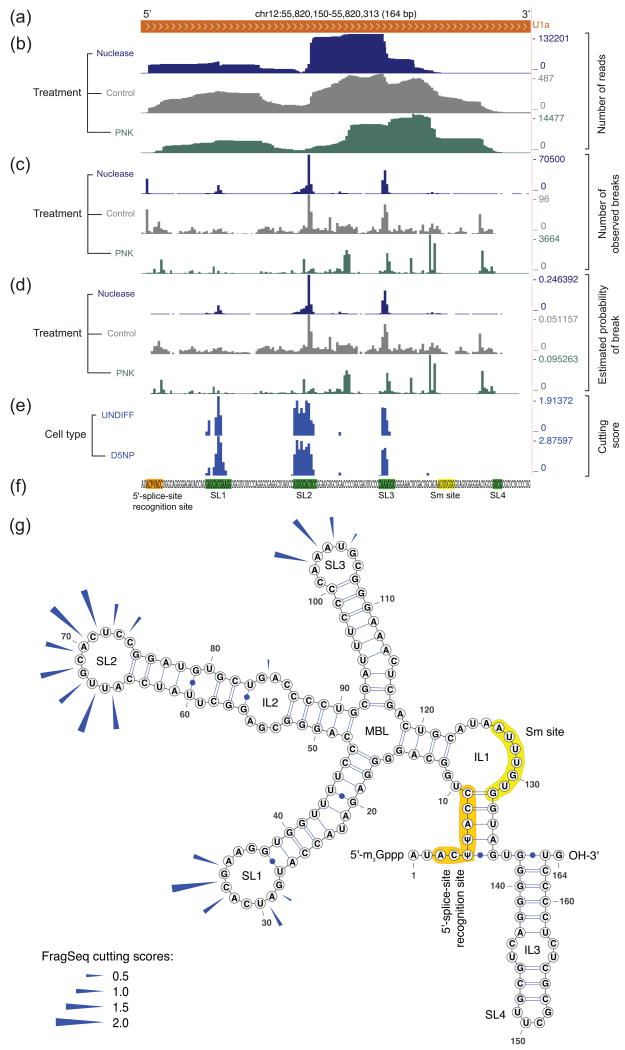
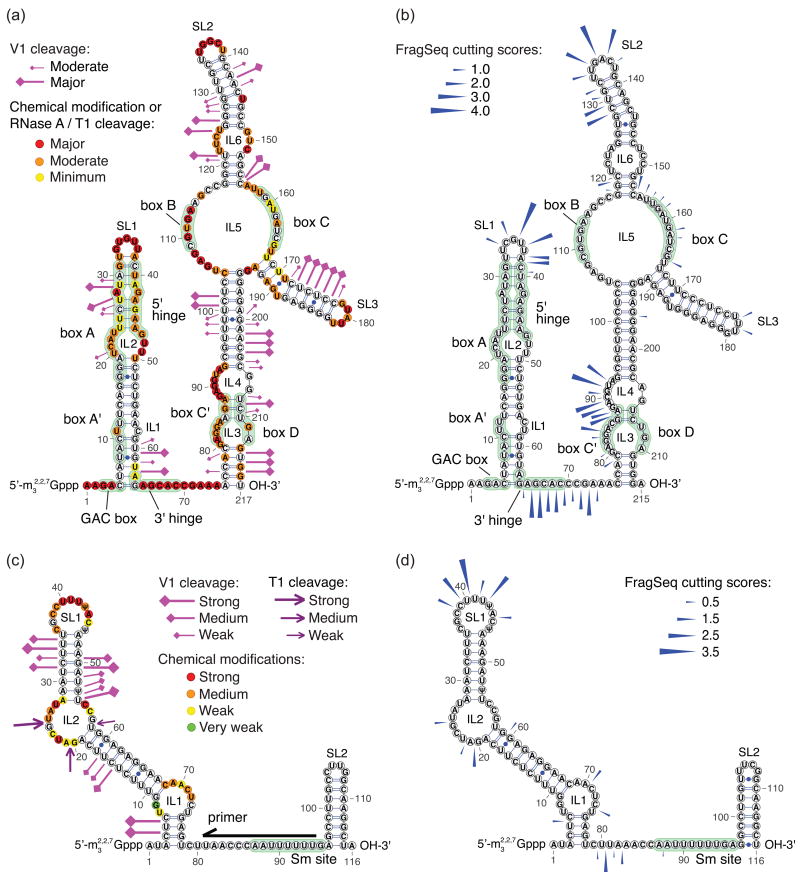
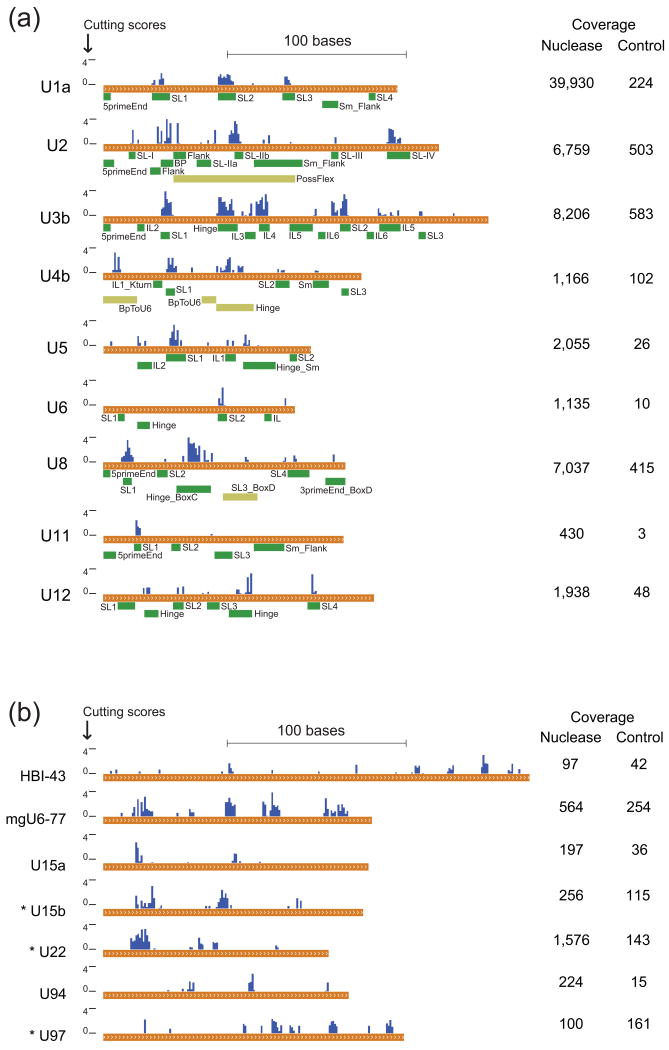
Classical approaches to determine structures of noncoding RNA (ncRNA) probed only one RNA at a time with enzymes and chemicals, using gel electrophoresis to identify reactive positions. To accelerate RNA structure inference, we developed fragmentation sequencing (FragSeq), a high-throughput RNA structure probing method that uses high-throughput RNA sequencing of fragments generated by digestion with nuclease P1, which specifically cleaves single-stranded nucleic acids. In experiments probing the entire mouse nuclear transcriptome, we accurately and simultaneously mapped single-stranded RNA regions in multiple ncRNAs with known structure. We probed in two cell types to verify reproducibility. We also identified and experimentally validated structured regions in ncRNAs with, to our knowledge, no previously reported probing data.
Figures
References
-
- Gesteland R, Cech T, Atkins J, editors. The RNA World. 3rd. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2005.
-
- Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. - PubMed
-
- Kapranov P, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
