

Molecular biology and pathology of prion strains in sporadic human prion diseases
- PMID: 21058033
- PMCID: PMC3077936
- DOI: 10.1007/s00401-010-0761-3
Molecular biology and pathology of prion strains in sporadic human prion diseases
Abstract
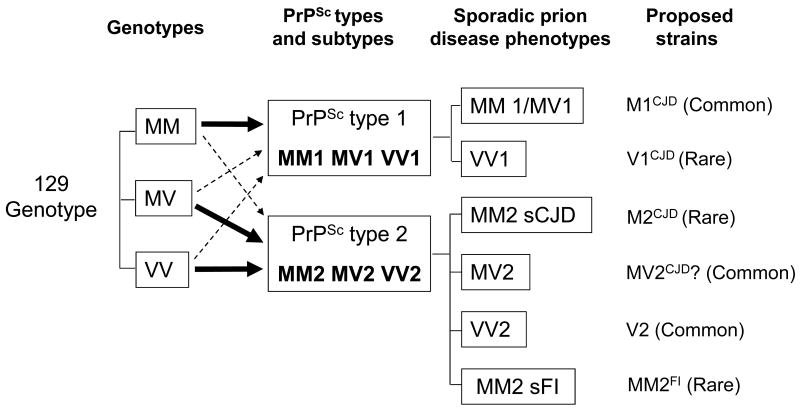
Prion diseases are believed to propagate by the mechanism involving self-perpetuating conformational conversion of the normal form of the prion protein, PrP(C), to the misfolded, pathogenic state, PrP(Sc). One of the most intriguing aspects of these disorders is the phenomenon of prion strains. It is believed that strain properties are fully encoded in distinct conformations of PrP(Sc). Strains are of practical relevance to human prion diseases as their diversity may explain the unusual heterogeneity of these disorders. The first insight into the molecular mechanisms underlying heterogeneity of human prion diseases was provided by the observation that two distinct disease phenotypes and their associated PrP(Sc) conformers co-distribute with distinct PrP genotypes as determined by the methionine/valine polymorphism at codon 129 of the PrP gene. Subsequent studies identified six possible combinations of the three genotypes (determined by the polymorphic codon 129) and two common PrP(Sc) conformers (named types 1 and 2) as the major determinants of the phenotype in sporadic human prion diseases. This scenario implies that each 129 genotype-PrP(Sc) type combination would be associated with a distinct disease phenotype and prion strain. However, notable exceptions have been found. For example, two genotype-PrP(Sc) type combinations are linked to the same phenotype, and conversely, the same combination was found to be associated with two distinct phenotypes. Furthermore, in some cases, PrP(Sc) conformers naturally associated with distinct phenotypes appear, upon transmission, to lose their phenotype-determining strain characteristics. Currently it seems safe to assume that typical sporadic prion diseases are associated with at least six distinct prion strains. However, the intrinsic characteristics that distinguish at least four of these strains remain to be identified.
Figures
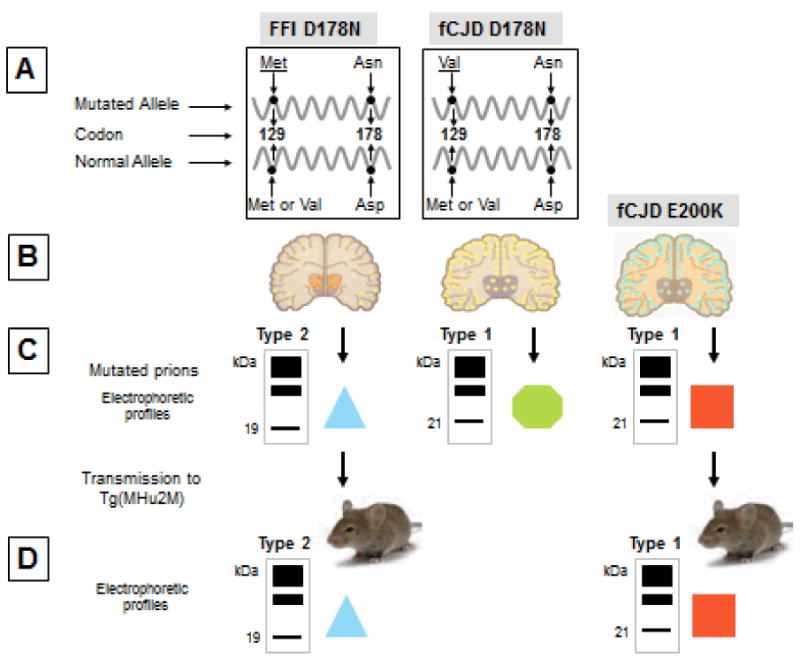
 : The light blue triangle represents the conformation of the PrPSc conformer from FFID178N;
: The light blue triangle represents the conformation of the PrPSc conformer from FFID178N;  : The green hexagon represents the conformation of PrPSc from fCJDD178N;
: The green hexagon represents the conformation of PrPSc from fCJDD178N;  : The red square represents the conformation of PrPSc from fCJDE200K which although being type one as fCJDD178N- associated PrPSc is presumably different based on the fCJDE200K distinct phenotype.
: The red square represents the conformation of PrPSc from fCJDE200K which although being type one as fCJDD178N- associated PrPSc is presumably different based on the fCJDE200K distinct phenotype.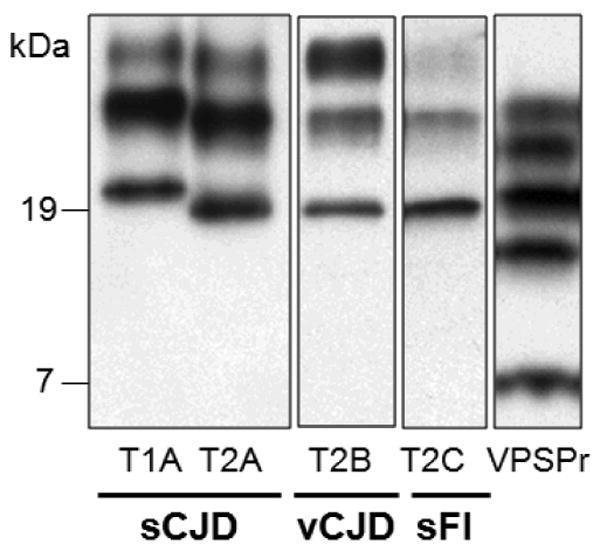
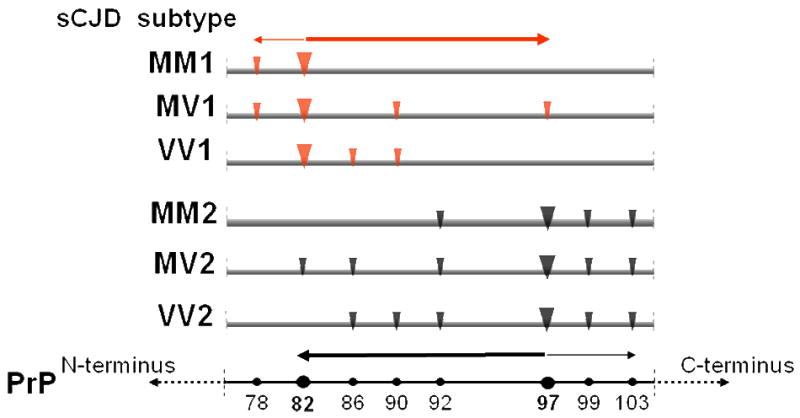
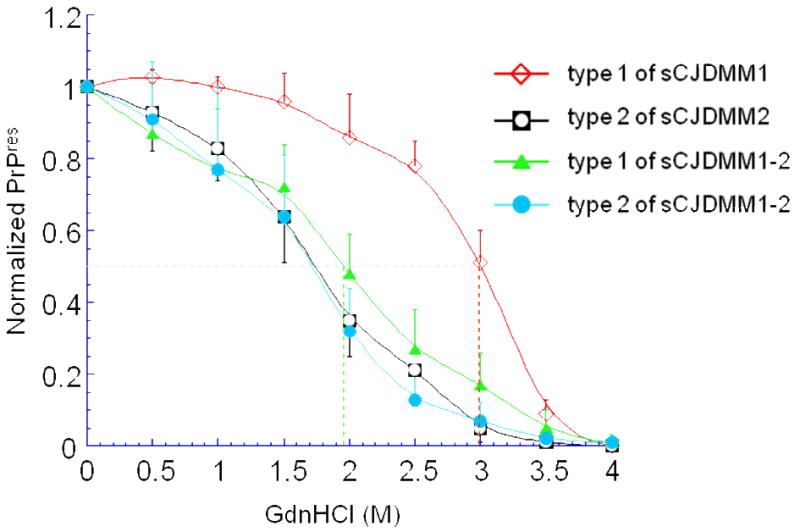
References
-
- Apetri AC, Vanik DL, Surewicz WK. Polymorphism at residue 129 modulates the conformational conversion of the D178N variant of human prion protein 90-231. Biochemistry. 2005;44:15880–15888. - PubMed
-
- Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol. 1992;73:329–334. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
