

Early host cell reactivation of an oxidatively damaged adenovirus-encoded reporter gene requires the Cockayne syndrome proteins CSA and CSB
- PMID: 21059811
- PMCID: PMC3044198
- DOI: 10.1093/mutage/geq096
Early host cell reactivation of an oxidatively damaged adenovirus-encoded reporter gene requires the Cockayne syndrome proteins CSA and CSB
Abstract
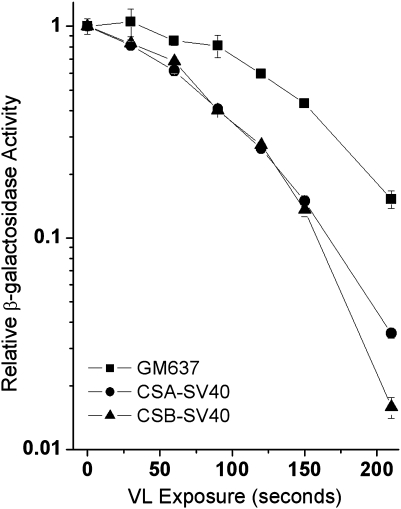
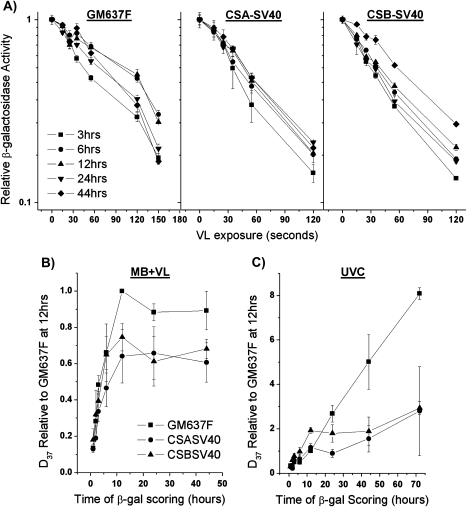
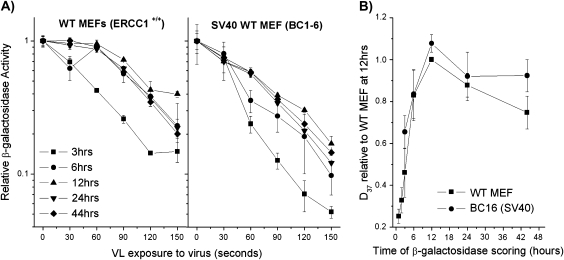
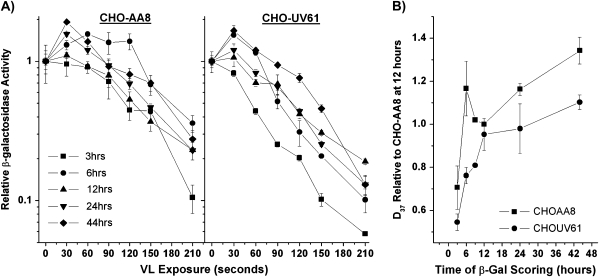
Reduced host cell reactivation (HCR) of a reporter gene containing 8-oxoguanine (8-oxoG) lesions in Cockayne syndrome (CS) fibroblasts has previously been attributed to increased 8-oxoG-mediated inhibition of transcription resulting from a deficiency in repair. This interpretation has been challenged by a report suggesting reduced expression from an 8-oxoG containing reporter gene occurs in all cells by a mechanism involving gene inactivation by 8-oxoG DNA glycosylase and this inactivation is strongly enhanced in the absence of the CS group B (CSB) protein. The observation of reduced gene expression in the absence of CSB protein led to speculation that decreased HCR in CS cells results from enhanced gene inactivation rather than reduced gene reactivation. Using an adenovirus-based β-galactosidase (β-gal) reporter gene assay, we have examined the effect of methylene blue plus visible light (MB + VL)-induced 8-oxoG lesions on the time course of gene expression in normal and CSA and CSB mutant human SV40-transformed fibroblasts, repair proficient and CSB mutant Chinese hamster ovary (CHO) cells and normal mouse embryo fibroblasts. We demonstrate that MB + VL treatment of the reporter leads to reduced expression of the damaged β-gal reporter relative to control at early time points following infection in all cells, consistent with in vivo inhibition of RNA polII-mediated transcription. In addition, we have demonstrated HCR of reporter gene expression occurs in all cell types examined. A significant reduction in the rate of gene reactivation in human SV40-transformed cells lacking functional CSA or CSB compared to normal cells was found. Similarly, a significant reduction in the rate of reactivation in CHO cells lacking functional CSB (CHO-UV61) was observed compared to the wild-type parental counterpart (CHO-AA8). The data presented demonstrate that expression of an oxidatively damaged reporter gene is reactivated over time and that CSA and CSB are required for normal reactivation.
Figures
Similar articles
-
Host cell reactivation of gene expression for an adenovirus-encoded reporter gene reflects the repair of UVC-induced cyclobutane pyrimidine dimers and methylene blue plus visible light-induced 8-oxoguanine.Mutagenesis. 2013 Sep;28(5):507-13. doi: 10.1093/mutage/get027. Epub 2013 Jun 21. Mutagenesis. 2013. PMID: 23793457 Free PMC article.
-
Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells.Nucleic Acids Res. 2002 Feb 1;30(3):782-93. doi: 10.1093/nar/30.3.782. Nucleic Acids Res. 2002. PMID: 11809892 Free PMC article.
-
Expression of an adenovirus encoded reporter gene and its reactivation following UVC and oxidative damage in cultured fish cells.Int J Radiat Biol. 2008 Jun;84(6):455-66. doi: 10.1080/09553000802078370. Int J Radiat Biol. 2008. PMID: 18470745
-
Repair of oxidatively generated DNA damage in Cockayne syndrome.Mech Ageing Dev. 2013 May-Jun;134(5-6):253-60. doi: 10.1016/j.mad.2013.03.001. Epub 2013 Mar 18. Mech Ageing Dev. 2013. PMID: 23518175 Review.
-
Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer.Cells. 2021 Apr 10;10(4):866. doi: 10.3390/cells10040866. Cells. 2021. PMID: 33920220 Free PMC article. Review.
Cited by
-
Mfd is required for rapid recovery of transcription following UV-induced DNA damage but not oxidative DNA damage in Escherichia coli.J Bacteriol. 2012 May;194(10):2637-45. doi: 10.1128/JB.06725-11. Epub 2012 Mar 16. J Bacteriol. 2012. PMID: 22427630 Free PMC article.
-
Host cell reactivation of gene expression for an adenovirus-encoded reporter gene reflects the repair of UVC-induced cyclobutane pyrimidine dimers and methylene blue plus visible light-induced 8-oxoguanine.Mutagenesis. 2013 Sep;28(5):507-13. doi: 10.1093/mutage/get027. Epub 2013 Jun 21. Mutagenesis. 2013. PMID: 23793457 Free PMC article.
References
-
- Neeley WL, Essigmann JM. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol. 2006;19:491–505. - PubMed
-
- Park EM, Shigenaga MK, Degan P, Korn TS, Kitzler JW, Wehr CM, Kolachana P, Ames BN. Assay of excised oxidative DNA lesions: isolation of 8-oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3375–3379. - PMC - PubMed
-
- Osterod M, Larsen E, Le Page F, Hengstler JG, Van Der Horst GT, Boiteux S, Klungland A, Epe B. A global DNA repair mechanism involving the Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene. 2002;21:8232–8239. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
