

Pigs and humans with cystic fibrosis have reduced insulin-like growth factor 1 (IGF1) levels at birth
- PMID: 21059918
- PMCID: PMC2996661
- DOI: 10.1073/pnas.1015281107
Pigs and humans with cystic fibrosis have reduced insulin-like growth factor 1 (IGF1) levels at birth
Abstract
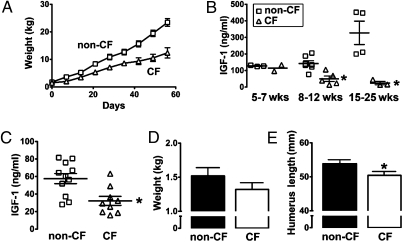
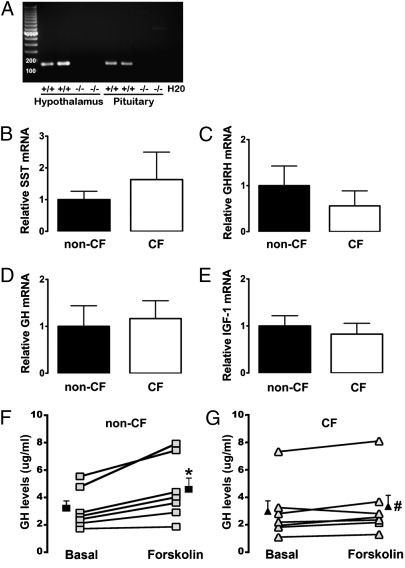
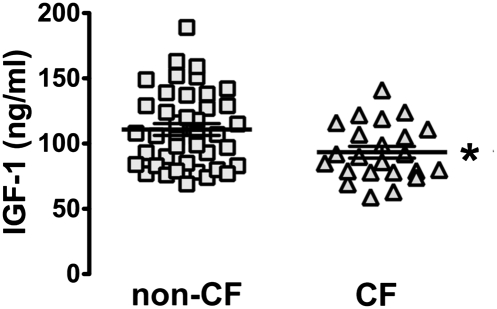
People with cystic fibrosis (CF) exhibit growth defects. That observation has been attributed, in part, to decreased insulin-like growth factor 1 (IGF1) levels, and the reduction has been blamed on malnutrition and pulmonary inflammation. However, patients with CF already have a reduced weight at birth, a manifestation not likely secondary to poor nutrition or inflammation. We found that, like humans, CF pigs were smaller than non-CF littermates and had lower IGF1 levels. To better understand the basis of IGF1 reduction, we studied newborn pigs and found low IGF1 levels within 12 h of birth. Moreover, humerus length and bone mineral content were decreased, consistent with less IGF1 activity in utero. These findings led us to test newborn humans with CF, and we found that they also had reduced IGF1 levels. Discovering lower IGF1 levels in newborn pigs and humans indicates that the decrease is not solely a consequence of malnutrition or pulmonary inflammation and that loss of cystic fibrosis transmembrane conductance regulator function has a more direct effect. Consistent with this hypothesis, we discovered reduced growth hormone release in organotypic pituitary slice cultures of newborn CF pigs. These findings may explain the long-standing observation that CF newborns are smaller than non-CF babies and why some patients with good clinical status fail to reach their growth potential. The results also suggest that measuring IGF1 levels might be of value as a biomarker to predict disease severity or the response to therapeutics. Finally, they raise the possibility that IGF1 supplementation beginning in infancy might be beneficial in CF.
Conflict of interest statement
Conflict of interest statement: Michael Welsh holds equity in Exemplar Genetics, which is licensing materials and technology related to this work.
Figures
References
-
- Corey M, McLaughlin FJ, Williams M, Levison H. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol. 1988;41:583–591. - PubMed
-
- Haeusler G, Frisch H, Waldhör T, Götz M. Perspectives of longitudinal growth in cystic fibrosis from birth to adult age. Eur J Pediatr. 1994;153:158–163. - PubMed
-
- Hardin DS, et al. A retrospective study of growth hormone use in adolescents with cystic fibrosis. Clin Endocrinol (Oxf) 2005;62:560–566. - PubMed
-
- Bronstein MN, et al. Pancreatic insufficiency, growth, and nutrition in infants identified by newborn screening as having cystic fibrosis. J Pediatr. 1992;120:533–540. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
