

Deficits in axonal transport precede ALS symptoms in vivo
- PMID: 21059924
- PMCID: PMC2996651
- DOI: 10.1073/pnas.1006869107
Deficits in axonal transport precede ALS symptoms in vivo
Abstract
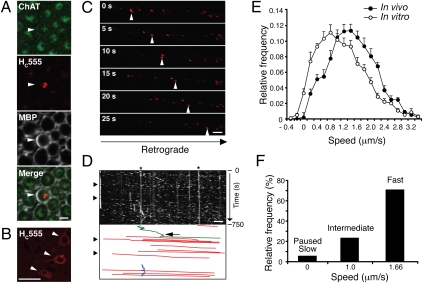
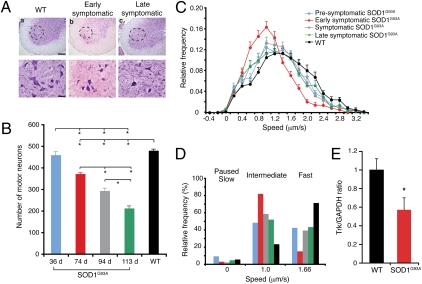
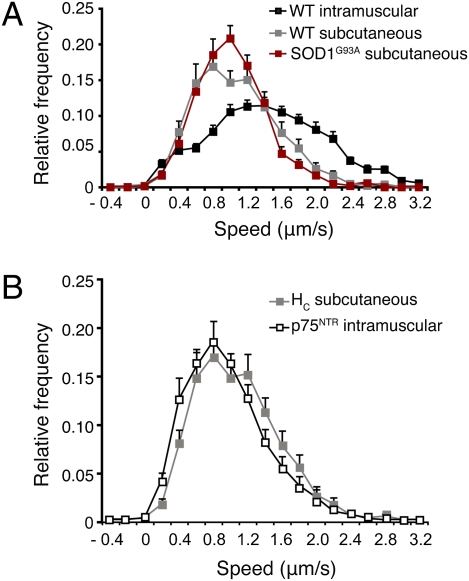
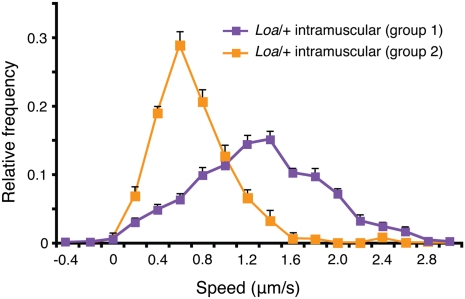
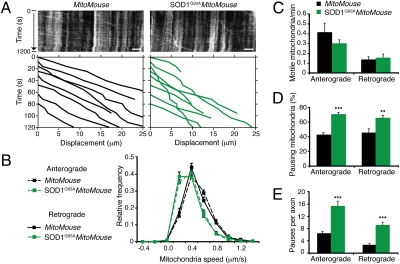
ALS is a fatal neurodegenerative disease characterized by selective motor neuron death resulting in muscle paralysis. Mutations in superoxide dismutase 1 (SOD1) are responsible for a subset of familial cases of ALS. Although evidence from transgenic mice expressing human mutant SOD1(G93A) suggests that axonal transport defects may contribute to ALS pathogenesis, our understanding of how these relate to disease progression remains unclear. Using an in vivo assay that allows the characterization of axonal transport in single axons in the intact sciatic nerve, we have identified clear axonal transport deficits in presymptomatic mutant mice. An impairment of axonal retrograde transport may therefore represent one of the earliest axonal pathologies in SOD1(G93A) mice, which worsens at an early symptomatic stage. A deficit in axonal transport may therefore be a key pathogenic event in ALS and an early disease indicator of motor neuron degeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Goldstein LS, Yang Z. Microtubule-based transport systems in neurons: The roles of kinesins and dyneins. Annu Rev Neurosci. 2000;23:39–71. - PubMed
-
- Chevalier-Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762:1094–1108. - PubMed
-
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
