

α-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8
- PMID: 21060150
- PMCID: PMC2993580
- DOI: 10.1172/JCI41196
α-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8
Abstract
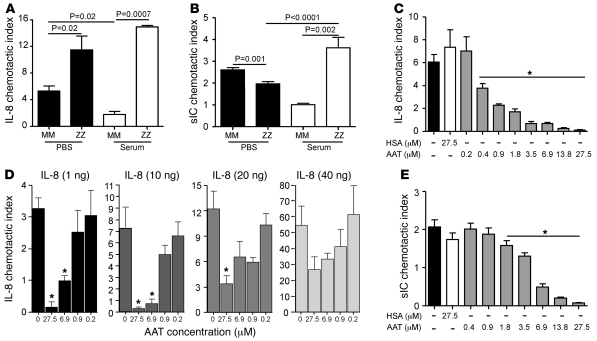
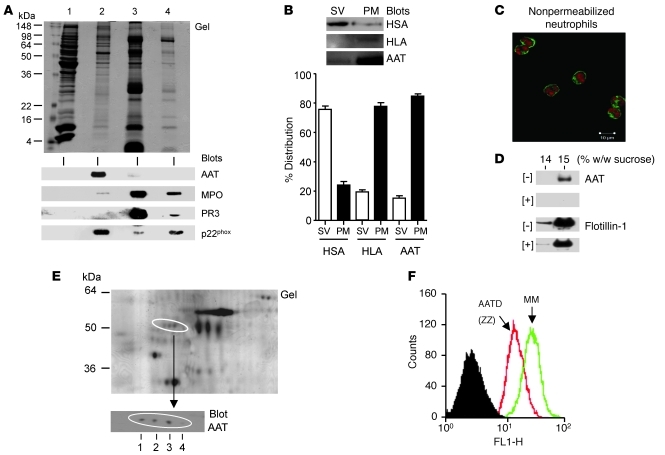
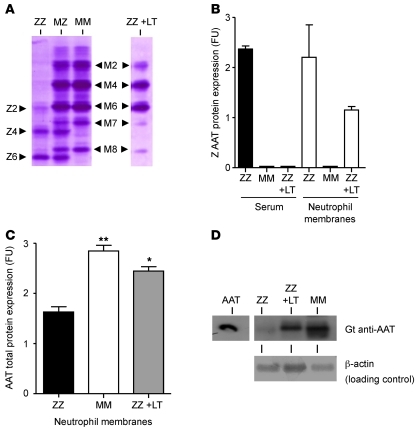
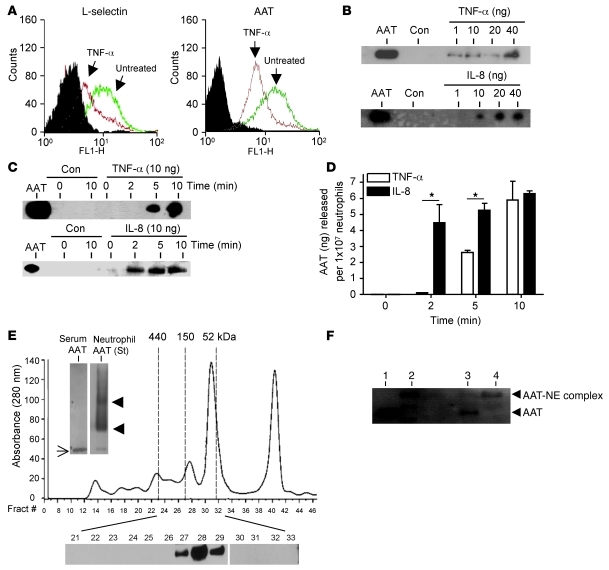
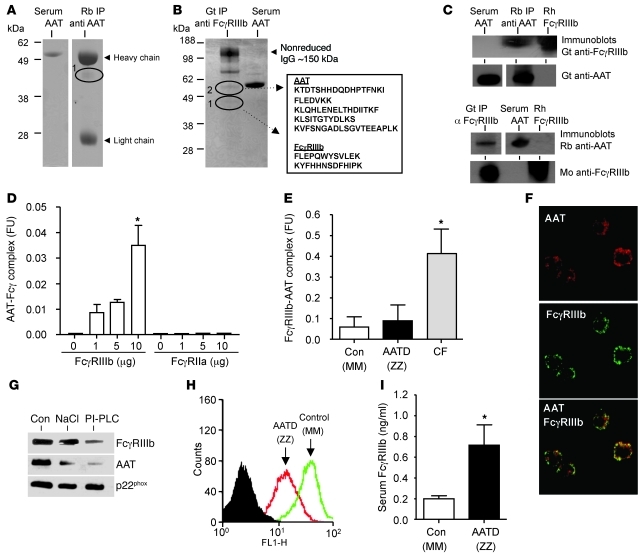
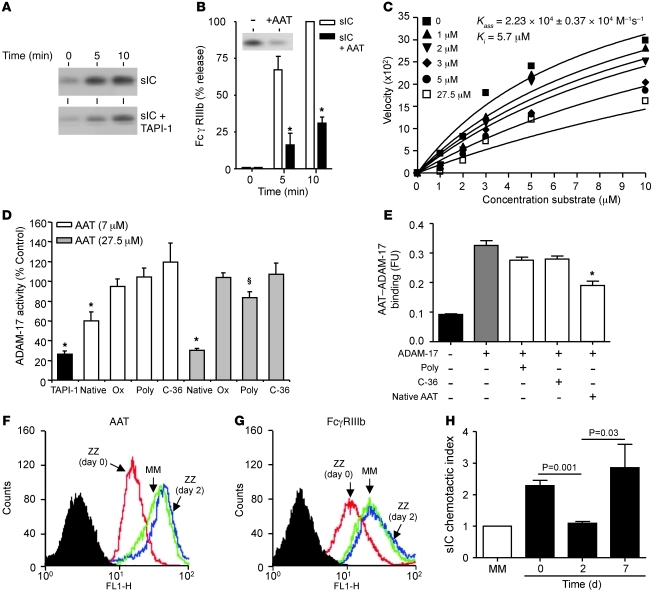
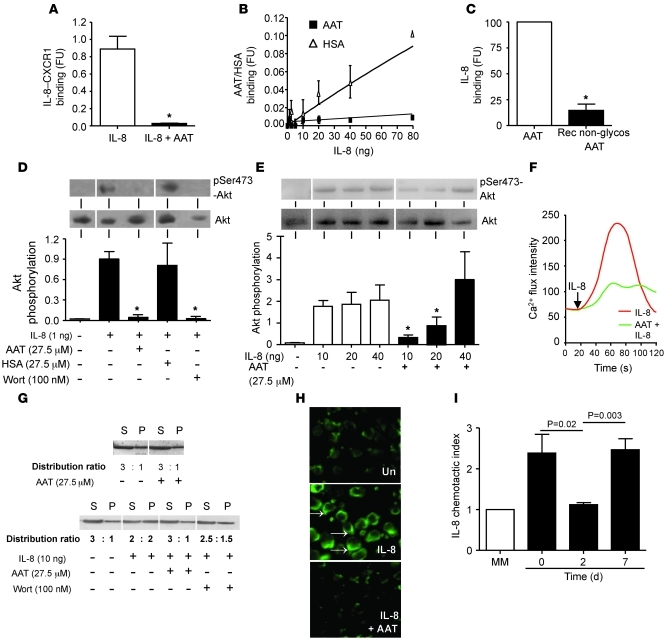
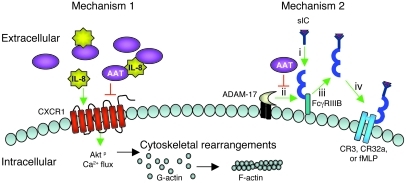
Hereditary deficiency of the protein α-1 antitrypsin (AAT) causes a chronic lung disease in humans that is characterized by excessive mobilization of neutrophils into the lung. However, the reason for the increased neutrophil burden has not been fully elucidated. In this study we have demonstrated using human neutrophils that serum AAT coordinates both CXCR1- and soluble immune complex (sIC) receptor-mediated chemotaxis by divergent pathways. We demonstrated that glycosylated AAT can bind to IL-8 (a ligand for CXCR1) and that AAT-IL-8 complex formation prevented IL-8 interaction with CXCR1. Second, AAT modulated neutrophil chemotaxis in response to sIC by controlling membrane expression of the glycosylphosphatidylinositol-anchored (GPI-anchored) Fc receptor FcγRIIIb. This process was mediated through inhibition of ADAM-17 enzymatic activity. Neutrophils isolated from clinically stable AAT-deficient patients were characterized by low membrane expression of FcγRIIIb and increased chemotaxis in response to IL-8 and sIC. Treatment of AAT-deficient individuals with AAT augmentation therapy resulted in increased AAT binding to IL-8, increased AAT binding to the neutrophil membrane, decreased FcγRIIIb release from the neutrophil membrane, and normalization of chemotaxis. These results provide new insight into the mechanism underlying the effect of AAT augmentation therapy in the pulmonary disease associated with AAT deficiency.
Figures
References
-
- Stockley RA, Shaw J, Afford SC, Morrison HM, Burnett D. Effect of alpha-1-proteinase inhibitor on neutrophil chemotaxis. Am J Respir Cell Mol Biol. 1990;2(2):163–170. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
