

State of the art and challenges in sequence based T-cell epitope prediction
- PMID: 21067545
- PMCID: PMC2981877
- DOI: 10.1186/1745-7580-6-S2-S3
State of the art and challenges in sequence based T-cell epitope prediction
Abstract
Sequence based T-cell epitope predictions have improved immensely in the last decade. From predictions of peptide binding to major histocompatibility complex molecules with moderate accuracy, limited allele coverage, and no good estimates of the other events in the antigen-processing pathway, the field has evolved significantly. Methods have now been developed that produce highly accurate binding predictions for many alleles and integrate both proteasomal cleavage and transport events. Moreover have so-called pan-specific methods been developed, which allow for prediction of peptide binding to MHC alleles characterized by limited or no peptide binding data. Most of the developed methods are publicly available, and have proven to be very useful as a shortcut in epitope discovery. Here, we will go through some of the history of sequence-based predictions of helper as well as cytotoxic T cell epitopes. We will focus on some of the most accurate methods and their basic background.
Figures
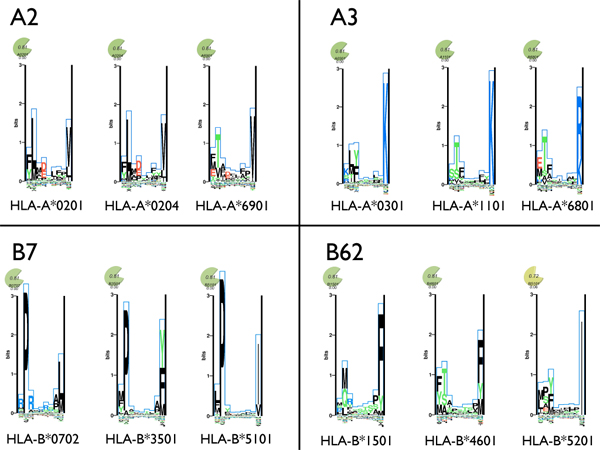
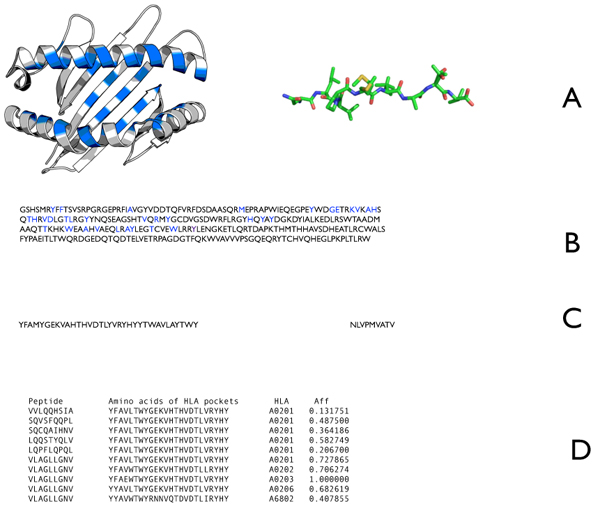
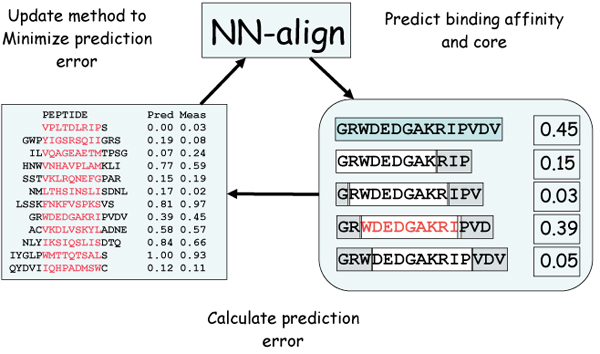
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials