

Oxidative stress induces reactivation of Kaposi's sarcoma-associated herpesvirus and death of primary effusion lymphoma cells
- PMID: 21068240
- PMCID: PMC3020037
- DOI: 10.1128/JVI.01742-10
Oxidative stress induces reactivation of Kaposi's sarcoma-associated herpesvirus and death of primary effusion lymphoma cells
Abstract
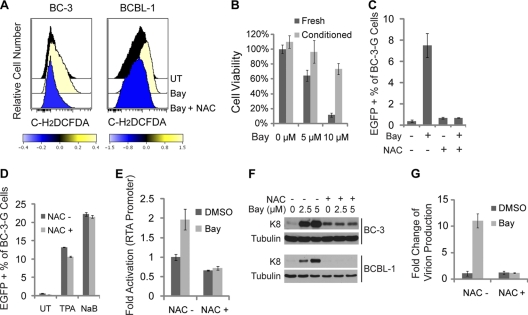
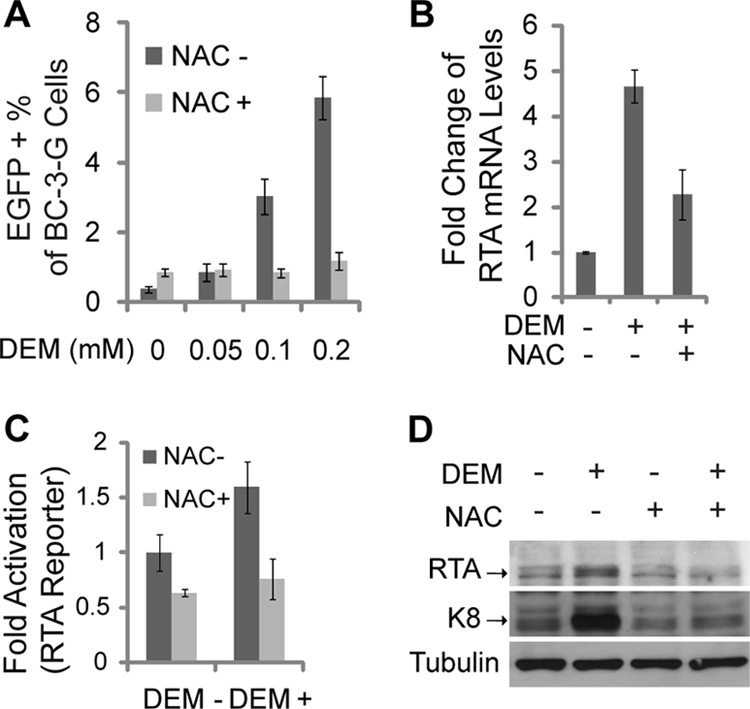
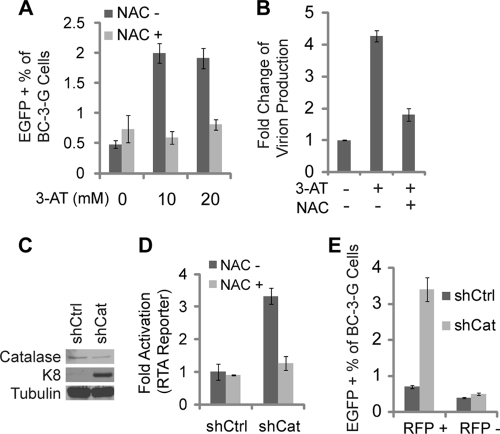
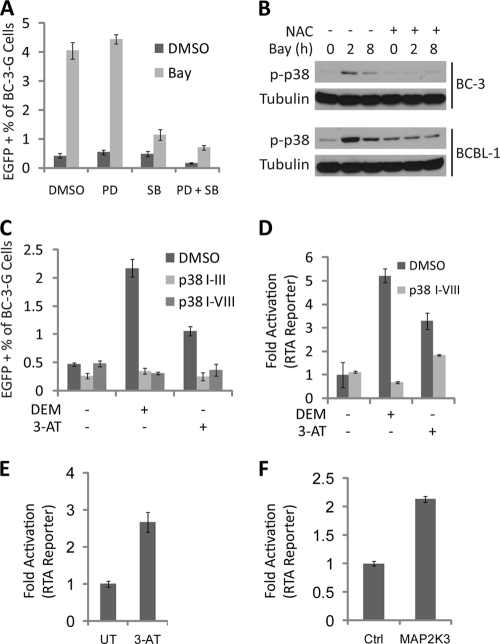
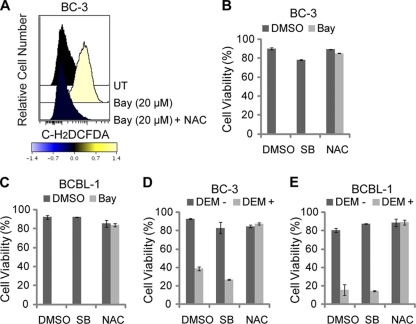
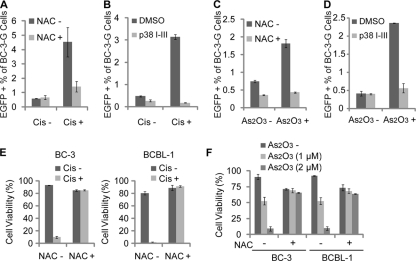
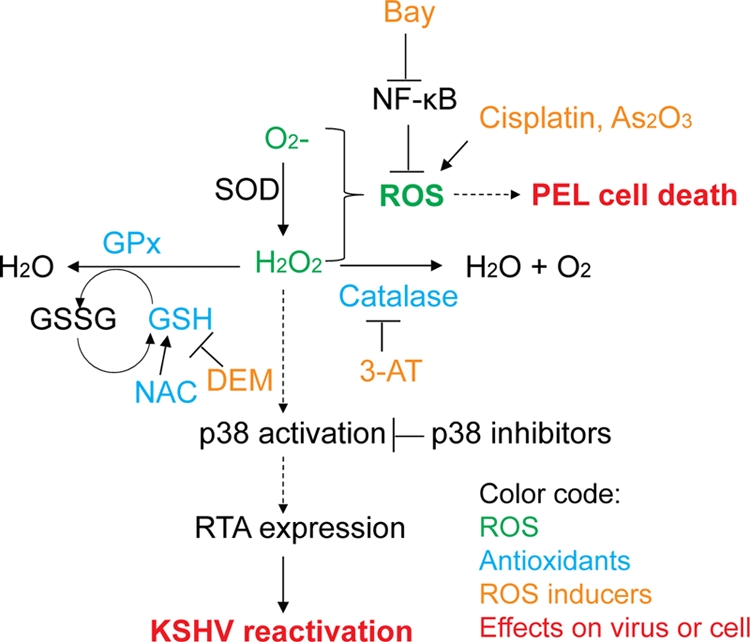
Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL) cells are predominantly infected with latent Kaposi's sarcoma-associated herpesvirus (KSHV), presenting a barrier to the destruction of tumor cells. Latent KSHV can be reactivated to undergo lytic replication. Here we report that in PEL cells, oxidative stress induced by upregulated reactive oxygen species (ROS) can lead to KSHV reactivation or cell death. ROS are upregulated by NF-κB inhibition and are required for subsequent KSHV reactivation. Disruption of the intracellular redox balance through depletion of the antioxidant glutathione or inhibition of the antioxidant enzyme catalase also induces KSHV reactivation, suggesting that hydrogen peroxide induces reactivation. In addition, p38 signaling is required for KSHV reactivation induced by ROS. Furthermore, treatment of PEL cells with a higher concentration of the NF-κB inhibitor than that used for inducing KSHV reactivation further upregulates ROS and induces massive cell death. ROS, but not p38 signaling, are required for PEL cell death induced by NF-κB inhibition as well as by glutathione depletion. Importantly, anticancer drugs, such as cisplatin and arsenic trioxide, also induce KSHV reactivation and PEL cell death in a ROS-dependent manner. Our study thus establishes a critical role for ROS and oxidative stress in the regulation of KSHV reactivation and PEL cell death. Disrupting the cellular redox balance may be a potential strategy for treating KSHV-associated lymphoma.
Figures
References
-
- Ambinder, R. F., K. D. Robertson, S. M. Moore, and J. Yang. 1996. Epstein-Barr virus as a therapeutic target in Hodgkin's disease and nasopharyngeal carcinoma. Semin. Cancer Biol. 7:217-226. - PubMed
-
- Brander, C., T. Suscovich, Y. Lee, P. T. Nguyen, P. O'Connor, J. Seebach, N. G. Jones, M. van Gorder, B. D. Walker, and D. T. Scadden. 2000. Impaired CTL recognition of cells latently infected with Kaposi's sarcoma-associated herpes virus. J. Immunol. 165:2077-2083. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
