

Amyloid-independent mechanisms in Alzheimer's disease pathogenesis
- PMID: 21068297
- PMCID: PMC3426835
- DOI: 10.1523/JNEUROSCI.4305-10.2010
Amyloid-independent mechanisms in Alzheimer's disease pathogenesis
Abstract
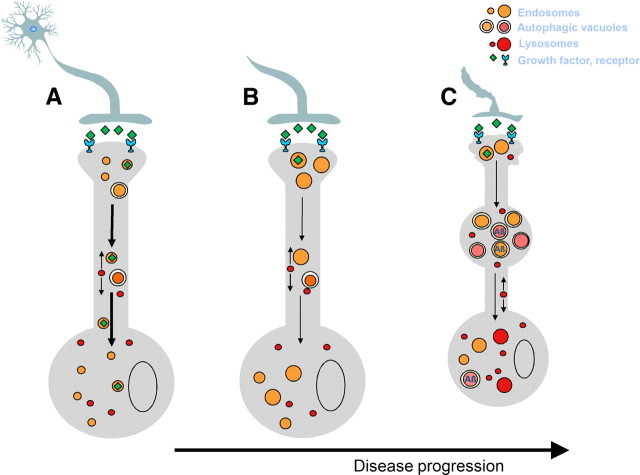
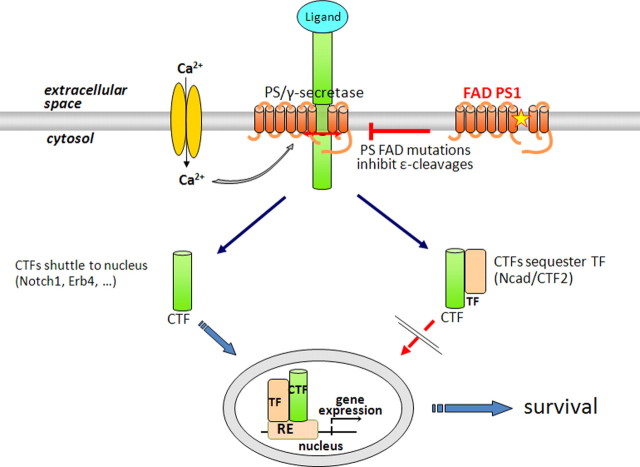
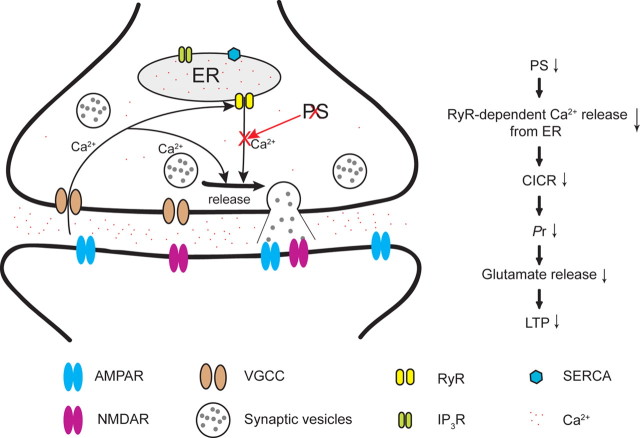
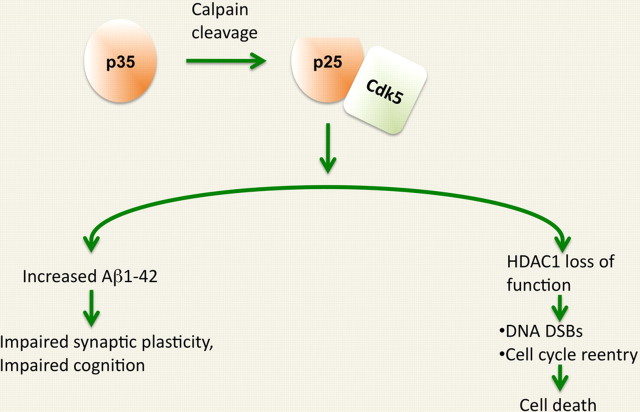
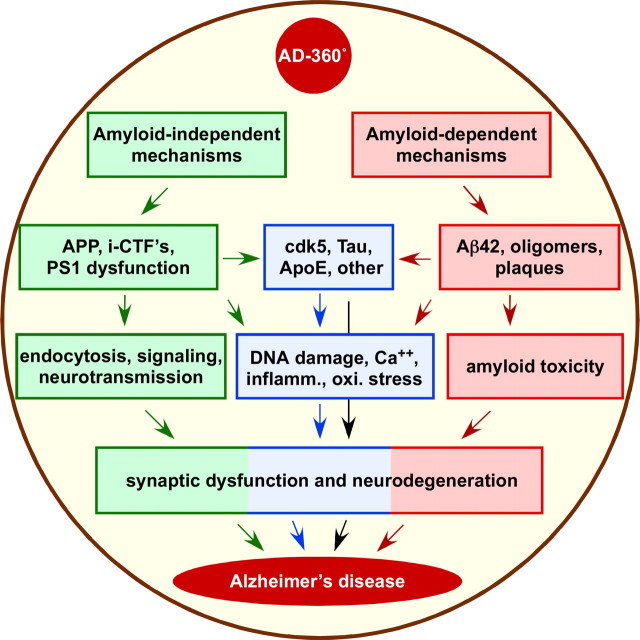
Despite the progress of the past two decades, the cause of Alzheimer's disease (AD) and effective treatments against it remain elusive. The hypothesis that amyloid-β (Aβ) peptides are the primary causative agents of AD retains significant support among researchers. Nonetheless, a growing body of evidence shows that Aβ peptides are unlikely to be the sole factor in AD etiology. Evidence that Aβ/amyloid-independent factors, including the actions of AD-related genes, also contribute significantly to AD pathogenesis was presented in a symposium at the 2010 Annual Meeting of the Society for Neuroscience. Here we summarize the studies showing how amyloid-independent mechanisms cause defective endo-lysosomal trafficking, altered intracellular signaling cascades, or impaired neurotransmitter release and contribute to synaptic dysfunction and/or neurodegeneration, leading to dementia in AD. A view of AD pathogenesis that encompasses both the amyloid-dependent and -independent mechanisms will help fill the gaps in our knowledge and reconcile the findings that cannot be explained solely by the amyloid hypothesis.
Figures
References
-
- Batelli S, Albani D, Prato F, Polito L, Franceschi M, Gavazzi A, Forloni G. Early-onset Alzheimer disease in an Italian family with presenilin-1 double mutation E318G and G394V. Alzheimer Dis Assoc Disord. 2008;22:184–187. - PubMed
-
- Beglopoulos V, Shen J. Regulation of CRE-dependent transcription by presenilins: prospects for therapy of Alzheimer's disease. Trends Pharmacol Sci. 2006;27:33–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01-AG026146/AG/NIA NIH HHS/United States
- R01 NS051874/NS/NINDS NIH HHS/United States
- RF1 NS041783/NS/NINDS NIH HHS/United States
- R01-NS041783/NS/NINDS NIH HHS/United States
- R56 AG008200/AG/NIA NIH HHS/United States
- P01-AG027916/AG/NIA NIH HHS/United States
- P01 AG017617/AG/NIA NIH HHS/United States
- P01-AG17617/AG/NIA NIH HHS/United States
- R01 AG008200/AG/NIA NIH HHS/United States
- R01 NS042818/NS/NINDS NIH HHS/United States
- P01 AG027916/AG/NIA NIH HHS/United States
- R01-NS042818/NS/NINDS NIH HHS/United States
- R01 AG026146/AG/NIA NIH HHS/United States
- R01 NS041783/NS/NINDS NIH HHS/United States
- AG008200/AG/NIA NIH HHS/United States
- R01-NS051874/NS/NINDS NIH HHS/United States
- R37 AG017926/AG/NIA NIH HHS/United States
- RF1 AG008200/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous