

Fatty acid amide hydrolase is a key regulator of endocannabinoid-induced myocardial tissue injury
- PMID: 21070851
- PMCID: PMC3022384
- DOI: 10.1016/j.freeradbiomed.2010.11.002
Fatty acid amide hydrolase is a key regulator of endocannabinoid-induced myocardial tissue injury
Abstract
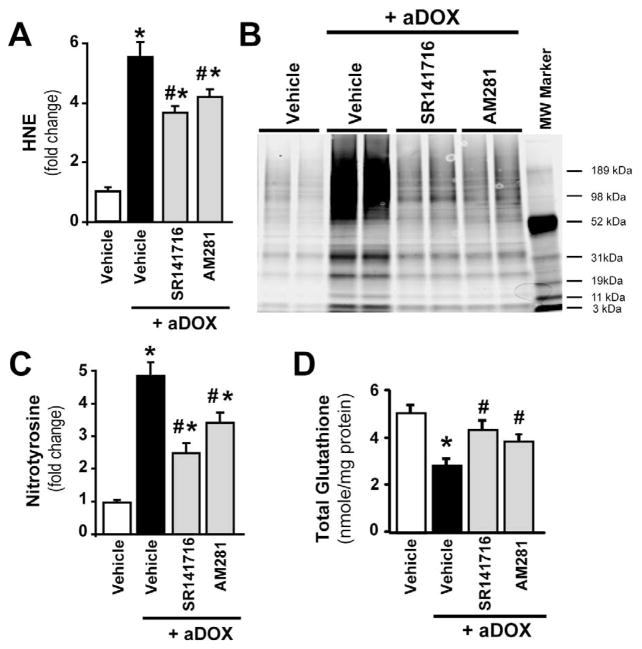
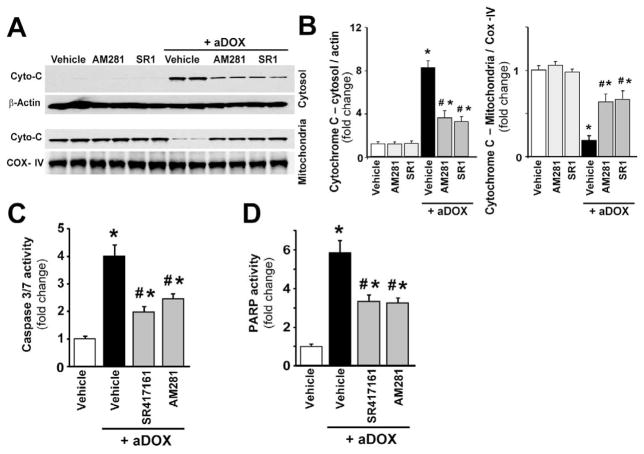
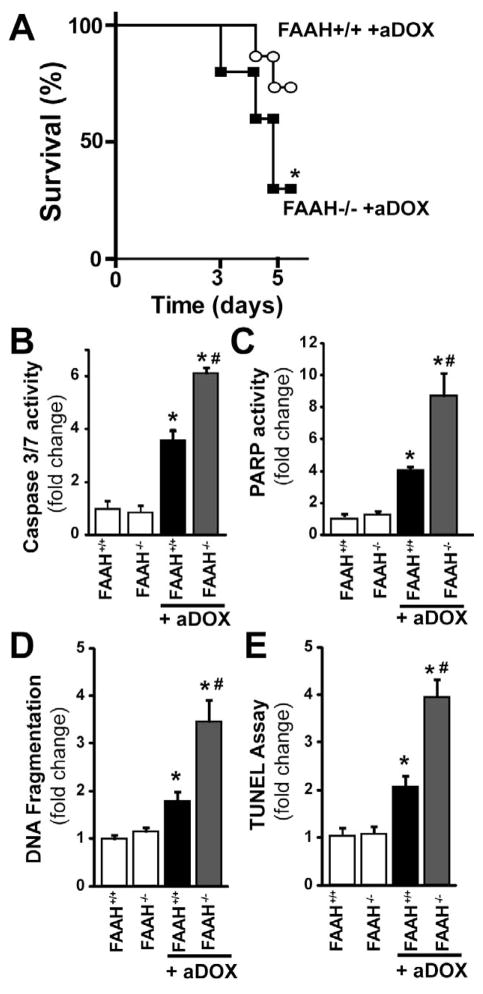
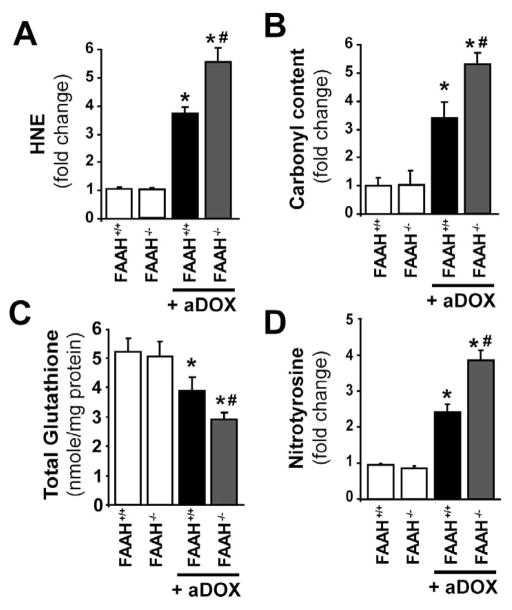
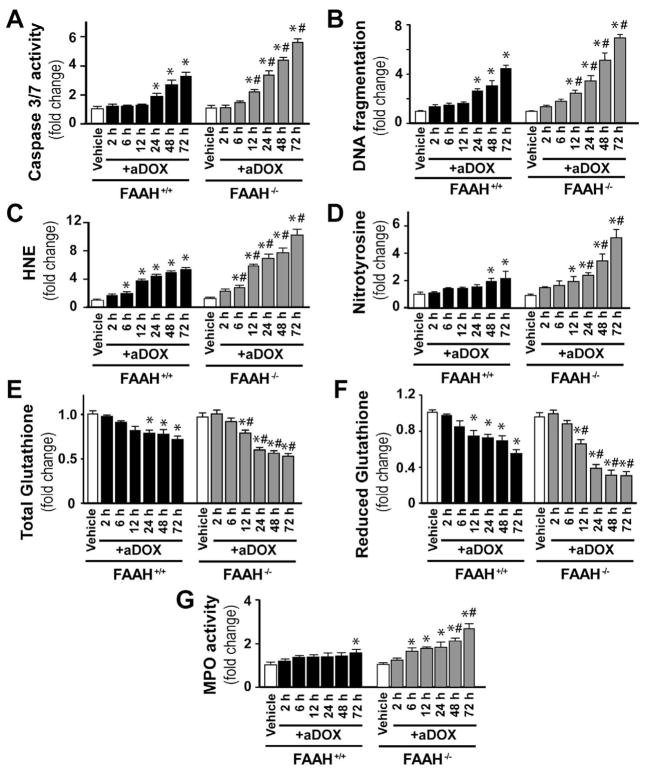
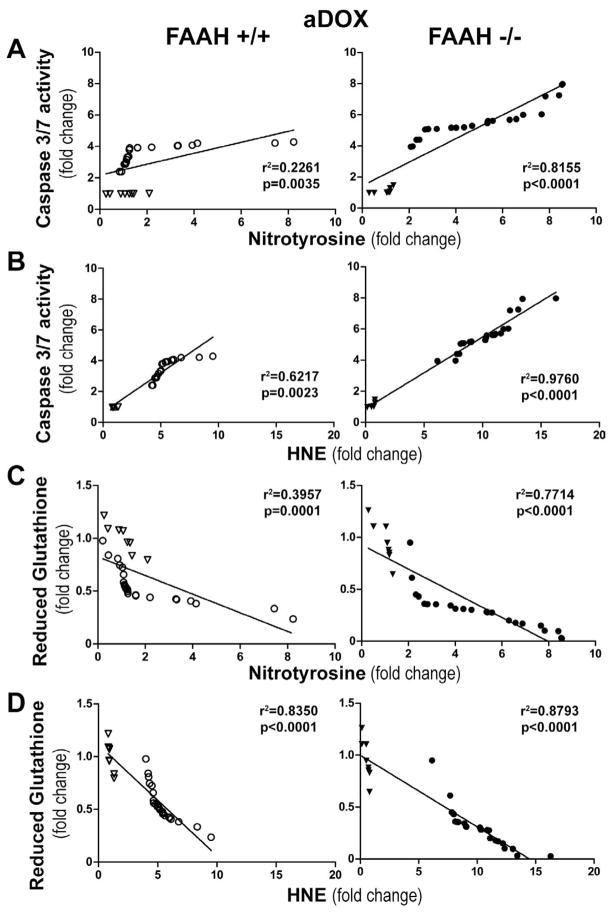
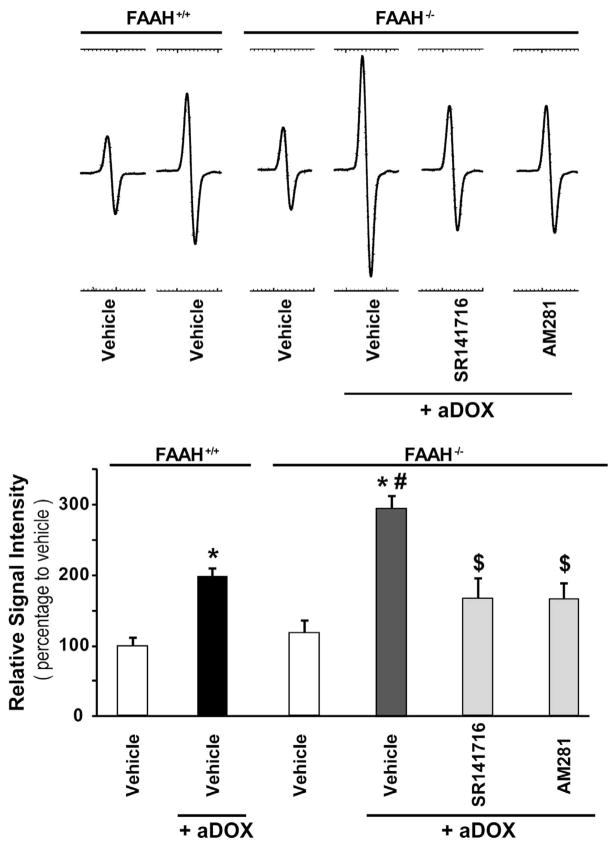
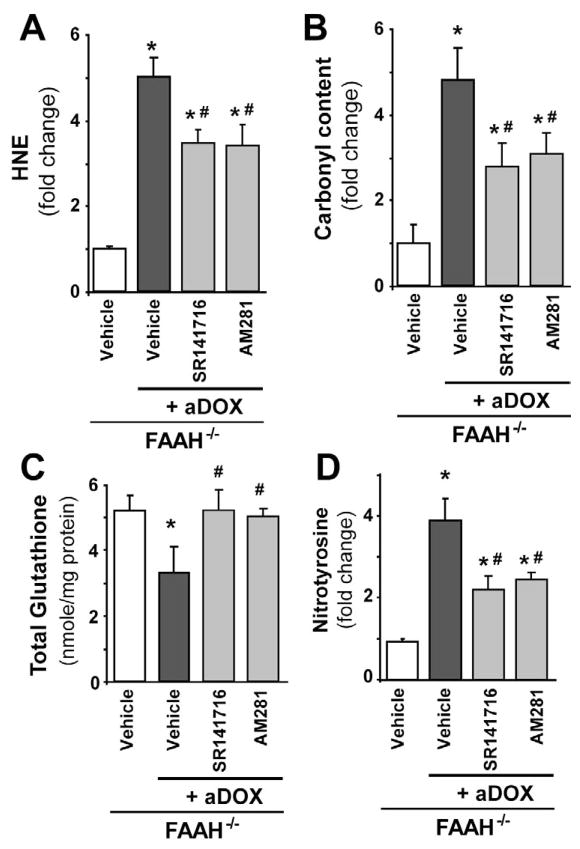
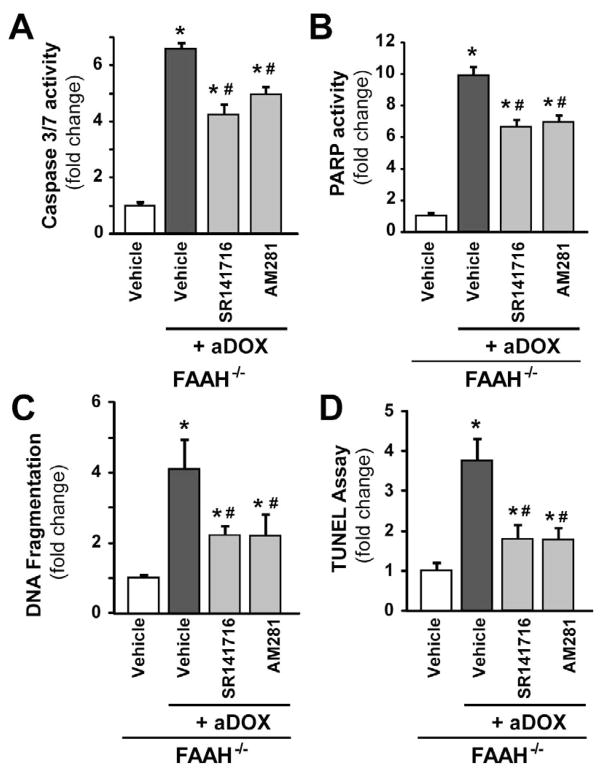
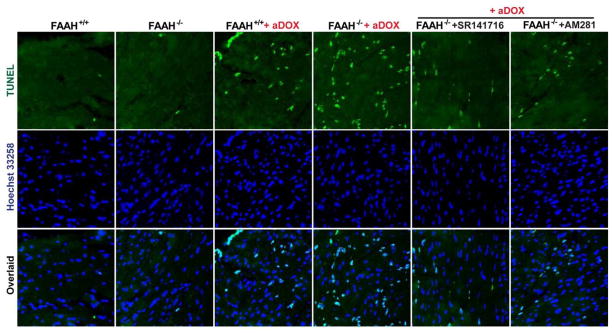
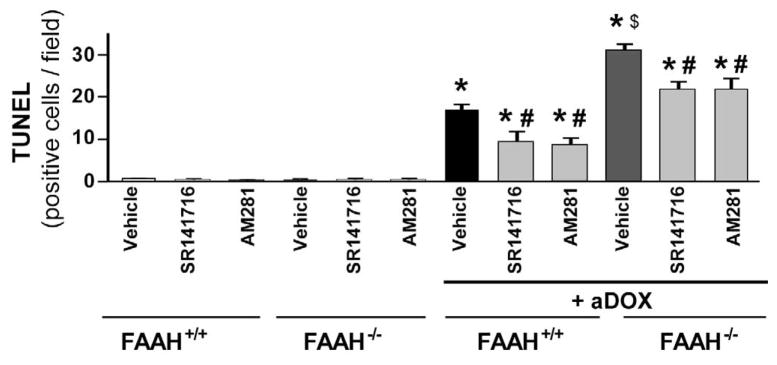
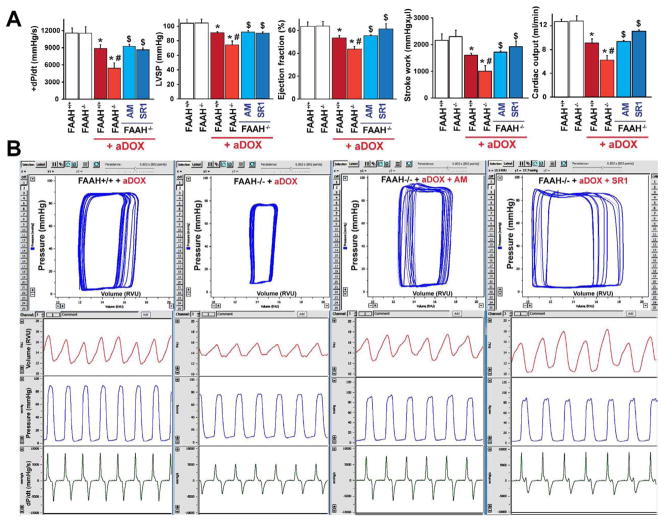
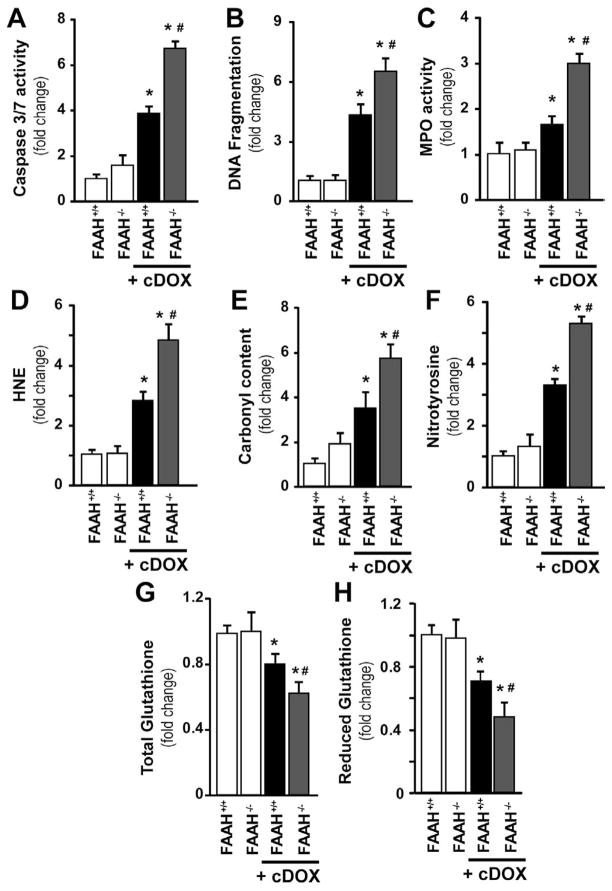
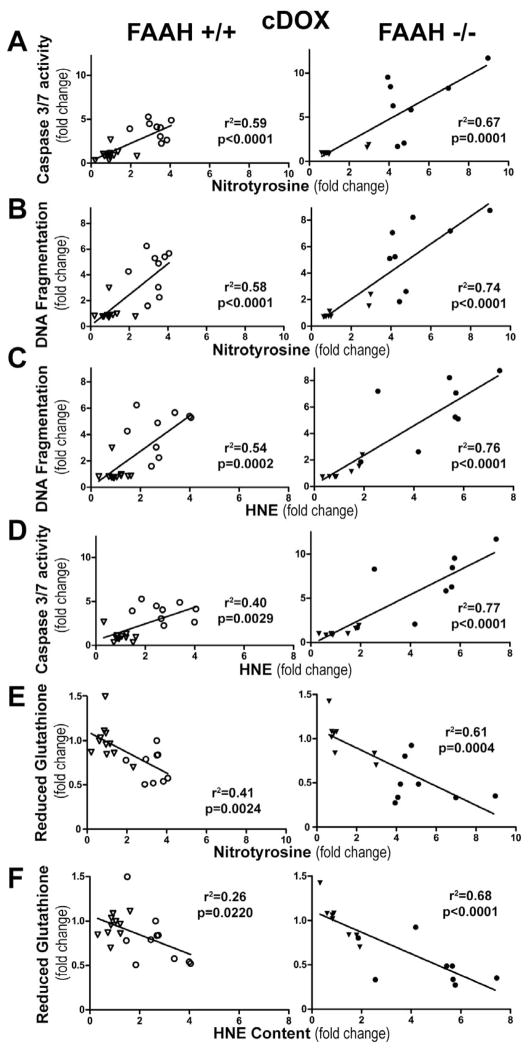
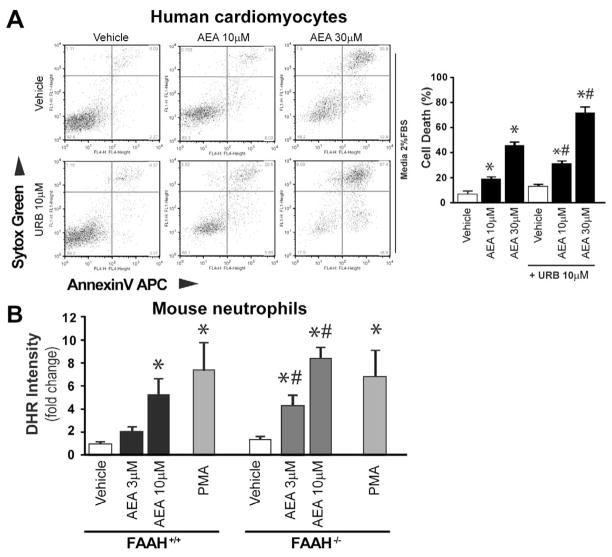
Previous studies have suggested that increased levels of endocannabinoids in various cardiovascular disorders (e.g., various forms of shock, cardiomyopathies, atherosclerosis) through the activation of CB(1) cannabinoid receptors may promote cardiovascular dysfunction and tissue injury. We have investigated the role of the main endocannabinoid anandamide-metabolizing enzyme (fatty acid amide hydrolase; FAAH) in myocardial injury induced by an important chemotherapeutic drug, doxorubicin (DOX; known for its cardiotoxicity mediated by increased reactive oxygen and nitrogen species generation), using well-established acute and chronic cardiomyopathy models in mice. The DOX-induced myocardial oxidative/nitrative stress (increased 4-hydroxynonenal, protein carbonyl, and nitrotyrosine levels and decreased glutathione content) correlated with multiple cell death markers, which were enhanced in FAAH knockout mice exhibiting significantly increased DOX-induced mortality and cardiac dysfunction compared to their wild type. The effects of DOX in FAAH knockouts were attenuated by CB(1) receptor antagonists. Furthermore, anandamide induced enhanced cell death in human cardiomyocytes pretreated with FAAH inhibitor and enhanced sensitivity to ROS generation in inflammatory cells of FAAH knockouts. These results suggest that in pathological conditions associated with acute oxidative/nitrative stress FAAH plays a key role in controlling the tissue injury that is, at least in part, mediated by the activation of CB(1) receptors by endocannabinoids.
Published by Elsevier Inc.
Conflict of interest statement
Figures
Similar articles
-
CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes.Cardiovasc Res. 2010 Mar 1;85(4):773-84. doi: 10.1093/cvr/cvp369. Epub 2009 Nov 26. Cardiovasc Res. 2010. PMID: 19942623 Free PMC article.
-
Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression, and apoptosis in mice lacking fatty acid amide hydrolase.Am J Physiol Heart Circ Physiol. 2007 Aug;293(2):H909-18. doi: 10.1152/ajpheart.00373.2007. Epub 2007 Apr 13. Am J Physiol Heart Circ Physiol. 2007. PMID: 17434980 Free PMC article.
-
Hemodynamic profile, responsiveness to anandamide, and baroreflex sensitivity of mice lacking fatty acid amide hydrolase.Am J Physiol Heart Circ Physiol. 2005 Aug;289(2):H533-41. doi: 10.1152/ajpheart.00107.2005. Epub 2005 Apr 8. Am J Physiol Heart Circ Physiol. 2005. PMID: 15821037 Free PMC article.
-
Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system.Curr Opin Chem Biol. 2003 Aug;7(4):469-75. doi: 10.1016/s1367-5931(03)00079-6. Curr Opin Chem Biol. 2003. PMID: 12941421 Review.
-
Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation.AAPS J. 2009 Mar;11(1):39-44. doi: 10.1208/s12248-008-9075-y. Epub 2009 Jan 29. AAPS J. 2009. PMID: 19184452 Free PMC article. Review.
Cited by
-
The Role of the Endocannabinoid System in the Brain-Gut Axis.Gastroenterology. 2016 Aug;151(2):252-66. doi: 10.1053/j.gastro.2016.04.015. Epub 2016 Apr 29. Gastroenterology. 2016. PMID: 27133395 Free PMC article. Review.
-
Metabolomic Studies of Tissue Injury in Nonhuman Primates Exposed to Gamma-Radiation.Int J Mol Sci. 2019 Jul 9;20(13):3360. doi: 10.3390/ijms20133360. Int J Mol Sci. 2019. PMID: 31323921 Free PMC article.
-
Drug-induced mitochondrial dysfunction and cardiotoxicity.Am J Physiol Heart Circ Physiol. 2015 Nov;309(9):H1453-67. doi: 10.1152/ajpheart.00554.2015. Epub 2015 Sep 18. Am J Physiol Heart Circ Physiol. 2015. PMID: 26386112 Free PMC article. Review.
-
Fatty Acid Amide Hydrolase Deficiency Is Associated with Deleterious Cardiac Effects after Myocardial Ischemia and Reperfusion in Mice.Int J Mol Sci. 2022 Oct 21;23(20):12690. doi: 10.3390/ijms232012690. Int J Mol Sci. 2022. PMID: 36293543 Free PMC article.
-
Activation of the endocannabinoid system mediates cardiac hypertrophy induced by rosiglitazone.Acta Pharmacol Sin. 2022 Sep;43(9):2302-2312. doi: 10.1038/s41401-022-00858-x. Epub 2022 Feb 21. Acta Pharmacol Sin. 2022. PMID: 35190698 Free PMC article.
References
-
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
