

Engagement of arginine finger to ATP triggers large conformational changes in NtrC1 AAA+ ATPase for remodeling bacterial RNA polymerase
- PMID: 21070941
- PMCID: PMC3001195
- DOI: 10.1016/j.str.2010.08.018
Engagement of arginine finger to ATP triggers large conformational changes in NtrC1 AAA+ ATPase for remodeling bacterial RNA polymerase
Abstract
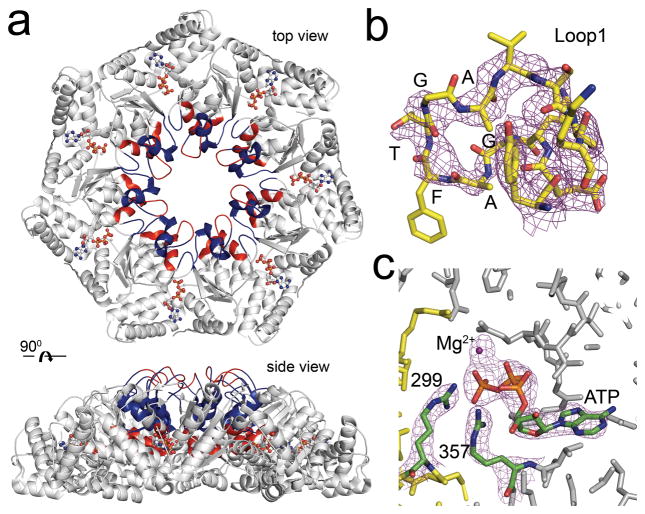
The NtrC-like AAA+ ATPases control virulence and other important bacterial activities through delivering mechanical work to σ54-RNA polymerase to activate transcription from σ54-dependent genes. We report the first crystal structure for such an ATPase, NtrC1 of Aquifex aeolicus, in which the catalytic arginine engages the γ-phosphate of ATP. Comparing the new structure with those previously known for apo and ADP-bound states supports a rigid-body displacement model that is consistent with large-scale conformational changes observed by low-resolution methods. First, the arginine finger induces rigid-body roll, extending surface loops above the plane of the ATPase ring to bind σ54. Second, ATP hydrolysis permits Pi release and retraction of the arginine with a reversed roll, remodeling σ54-RNAP. This model provides a fresh perspective on how ATPase subunits interact within the ring-ensemble to promote transcription, directing attention to structural changes on the arginine-finger side of an ATP-bound interface.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Conflict of interest statement
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Figures







Comment in
-
An ATPase R-finger leaves its print on transcriptional activation.Structure. 2010 Nov 10;18(11):1391-2. doi: 10.1016/j.str.2010.10.002. Structure. 2010. PMID: 21070936
References
-
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges N, Pannu NS, et al. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallographica. 1998;D54:905–921. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
