

A critical role for chloride channel-3 (CIC-3) in smooth muscle cell activation and neointima formation
- PMID: 21071705
- PMCID: PMC3025755
- DOI: 10.1161/ATVBAHA.110.217604
A critical role for chloride channel-3 (CIC-3) in smooth muscle cell activation and neointima formation
Abstract
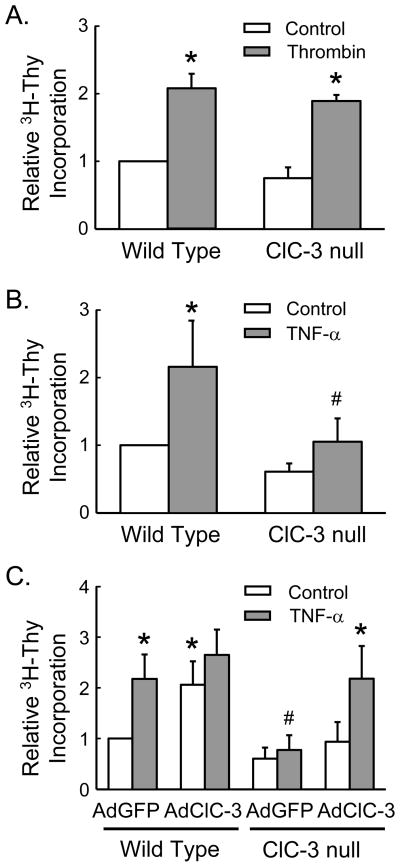
Objective: We have shown that the chloride-proton antiporter chloride channel-3 (ClC-3) is required for endosome-dependent signaling by the Nox1 NADPH oxidase in SMCs. In this study, we tested the hypothesis that ClC-3 is necessary for proliferation of smooth muscle cells (SMCs) and contributes to neointimal hyperplasia following vascular injury.
Methods and results: Studies were performed in SMCs isolated from the aorta of ClC-3-null and littermate control (wild-type [WT]) mice. Thrombin and tumor necrosis factor-α (TNF-α) each caused activation of both mitogen activated protein kinase extracellular signal-regulated kinases 1 and 2 and the matrix-degrading enzyme matrix metalloproteinase-9 and cell proliferation of WT SMCs. Whereas responses to thrombin were preserved in ClC-3-null SMCs, the responses to TNF-α were markedly impaired. These defects normalized following gene transfer of ClC-3. Carotid injury increased vascular ClC-3 expression, and compared with WT mice, ClC-3-null mice exhibited a reduction in neointimal area of the carotid artery 28 days after injury.
Conclusions: ClC-3 is necessary for the activation of SMCs by TNF-α but not thrombin. Deficiency of ClC-3 markedly reduces neointimal hyperplasia following vascular injury. In view of our previous findings, this observation is consistent with a role for ClC-3 in endosomal Nox1-dependent signaling. These findings identify ClC-3 as a novel target for the prevention of inflammatory and proliferative vascular diseases.
Figures





Comment in
-
Endosomal ClC-3 and Nox1: moving marksmen of redox signaling?Arterioscler Thromb Vasc Biol. 2011 Feb;31(2):240-2. doi: 10.1161/ATVBAHA.110.220053. Arterioscler Thromb Vasc Biol. 2011. PMID: 21248280 Free PMC article. No abstract available.
References
-
- Ross R. Atherosclerosis is an inflammatory disease. American Heart Journal. 1999;138:S419–S420. - PubMed
-
- Schwartz SM, deBlois D, O’Brien ERM. The intima. Soil for atherosclerosis and restenosis. Circulation Research. 1995;77:445–465. - PubMed
-
- Andrew C. Newby ABZ. Molecular mechanisms in intimal hyperplasia. The Journal of Pathology. 2000;190:300–309. - PubMed
-
- Kalmes A, Vesti BR, Daum G, Abraham JA, Clowes AW. Heparin Blockade of Thrombin-Induced Smooth Muscle Cell Migration Involves Inhibition of Epidermal Growth Factor (EGF) Receptor Transactivation by Heparin-Binding EGF-Like Growth Factor. Circ Res. 2000;87:92–98. - PubMed
-
- Darmoul D, Gratio V, Devaud H, Peiretti F, Laburthe M. Activation of proteinase-activated receptor 1 promotes human colon cancer cell proliferation through epidermal growth factor receptor transactivation. Molecular Cancer Research. 2004;2:514–522. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
