

Recent advances in macromolecular hydrodynamic modeling
- PMID: 21073955
- PMCID: PMC3085554
- DOI: 10.1016/j.ymeth.2010.10.005
Recent advances in macromolecular hydrodynamic modeling
Abstract
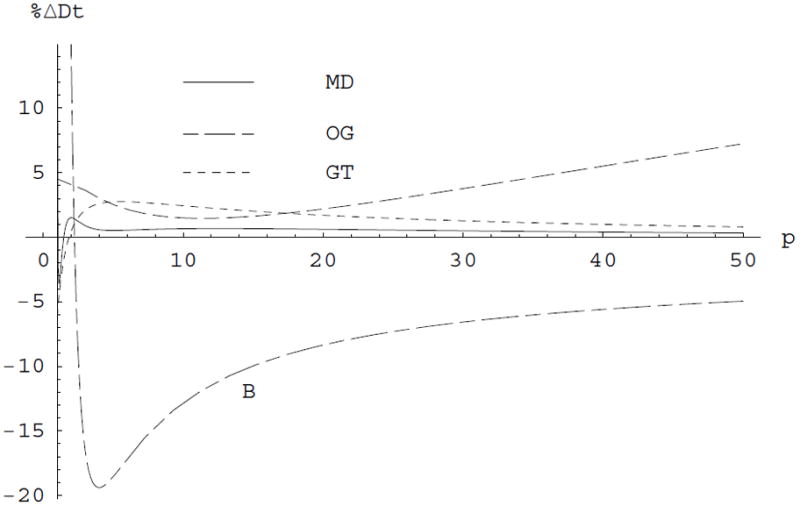
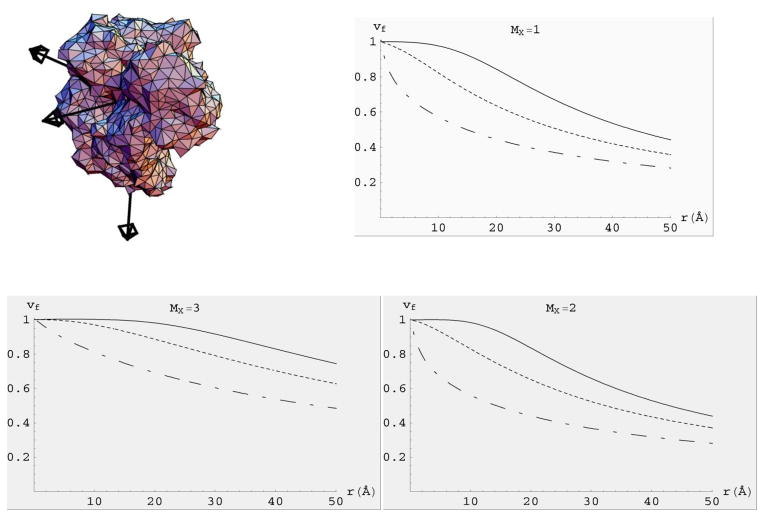
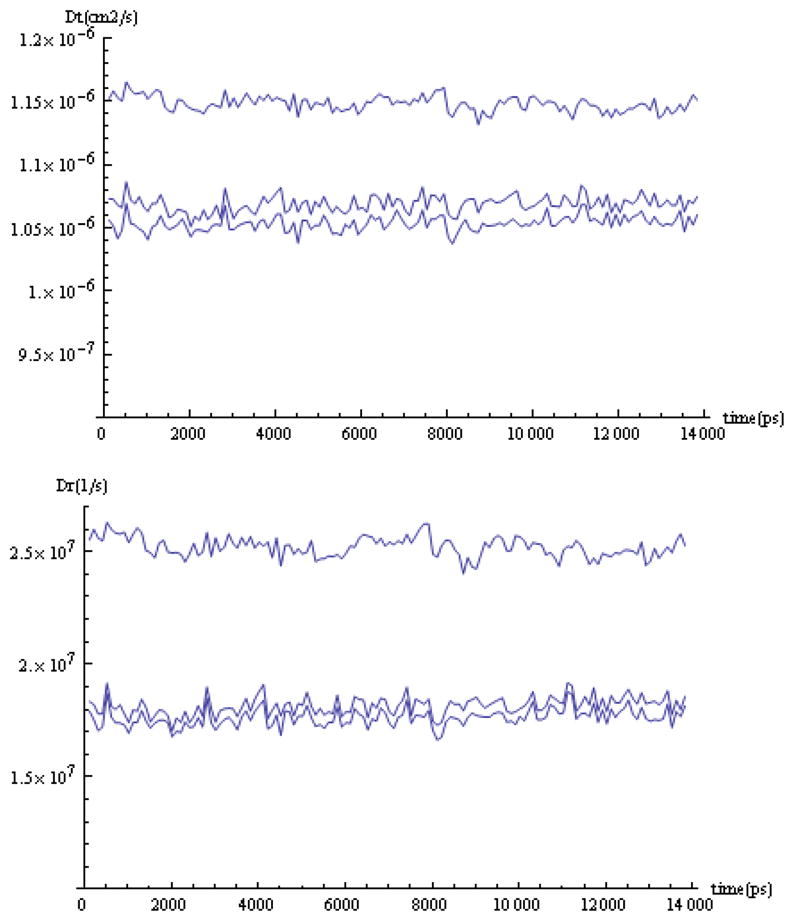
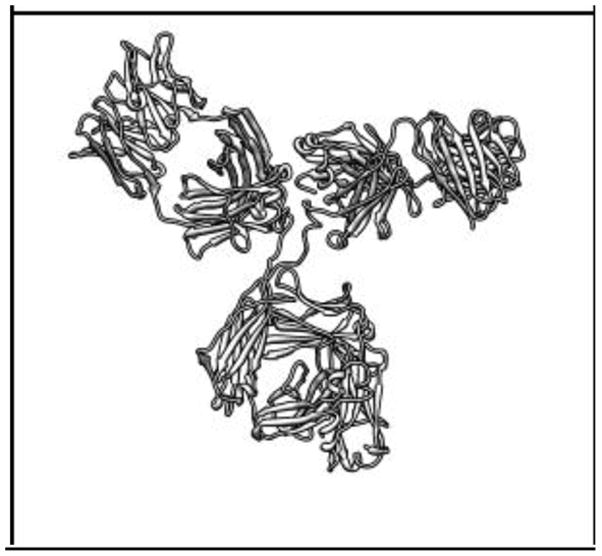
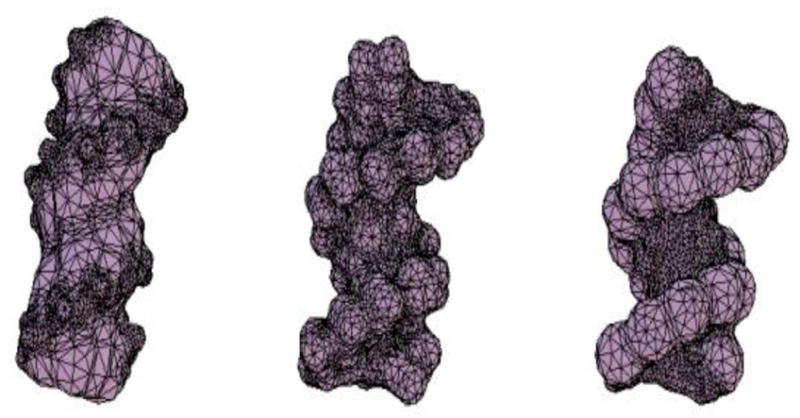
The modern implementation of the boundary element method [23] has ushered unprecedented accuracy and precision for the solution of the Stokes equations of hydrodynamics with stick boundary conditions. This article begins by reviewing computations with the program BEST of smooth surface objects such as ellipsoids, the dumbbell, and cylinders that demonstrate that the numerical solution of the integral equation formulation of hydrodynamics yields very high precision and accuracy. When BEST is used for macromolecular computations, the limiting factor becomes the definition of the molecular hydrodynamic surface and the implied effective solvation of the molecular surface. Studies on 49 different proteins, ranging in molecular weight from 9 to over 400kDa, have shown that a model using a 1.1Å thick hydration layer describes all protein transport properties very well for the overwhelming majority of them. In addition, this data implies that the crystal structure is an excellent representation of the average solution structure for most of them. In order to investigate the origin of a handful of significant discrepancies in some multimeric proteins (about -20% observed in the intrinsic viscosity), the technique of Molecular Dynamics simulation (MD) has been incorporated into the research program. A preliminary study of dimeric α-chymotrypsin using approximate implicit water MD is presented. In addition I describe the successful validation of modern protein force fields, ff03 and ff99SB, for the accurate computation of solution structure in explicit water simulation by comparison of trajectory ensemble average computed transport properties with experimental measurements. This work includes small proteins such as lysozyme, ribonuclease and ubiquitin using trajectories around 10ns duration. We have also studied a 150kDa flexible monoclonal IgG antibody, Trastuzumab, with multiple independent trajectories encompassing over 320ns of simulation. The close agreement within experimental error of the computed and measured properties allows us to conclude that MD does produce structures typical of those in solution, and that flexible molecules can be properly described using the method of ensemble averaging over a trajectory. We review similar work on the study of a transfer RNA molecule and DNA oligomers that demonstrate that within 3% a simple uniform hydration model 1.1Å thick provides agreement with experiment for these nucleic acids. In the case of linear oligomers, the precision can be improved close to 1% by a non-uniform hydration model that hydrates mainly in the DNA grooves, in agreement with high resolution X-ray diffraction. We conclude with a vista on planned improvements for the BEST program to decrease its memory requirements and increase its speed without sacrificing accuracy.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures


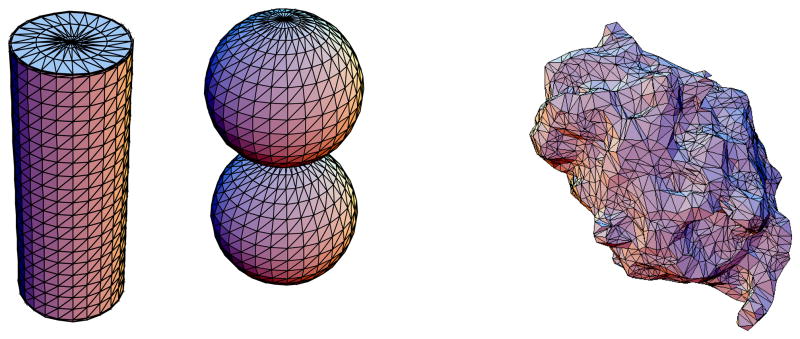


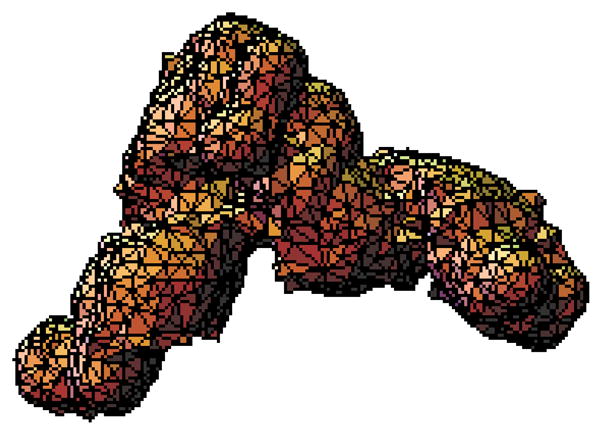

References
-
- Berne B, Pecora R. Dynamic Light Scattering: with applications to Chemistry, Biology and Physics. Wiley-Interscience; New York: 1976.
-
- Eden D, Elias JG. Transient Electric Birefringence of DNA restriction fragments and the filamentous virus Pf3. In: Dahneke B, editor. Measurement of Suspended Particles by Quasi-Elastic Light Scattering. Wiley-Interscience; New York: 1983.
-
- Stryer L. Fluorescence spectroscopy of proteins. Science. 1968;162:526–533. - PubMed
-
- Stallmach F, Galvosas P. Spin Echo NMR Diffusion Studies. Annual Reports on NMR Spectroscopy. 2007;61:51–131.
-
- Sanders JKM, Hunter BK. Modern NMR Spectroscopy. Oxford University Press; New York: 1987.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
