

Protein quality control during erythropoiesis and hemoglobin synthesis
- PMID: 21075281
- PMCID: PMC4136498
- DOI: 10.1016/j.hoc.2010.08.013
Protein quality control during erythropoiesis and hemoglobin synthesis
Abstract
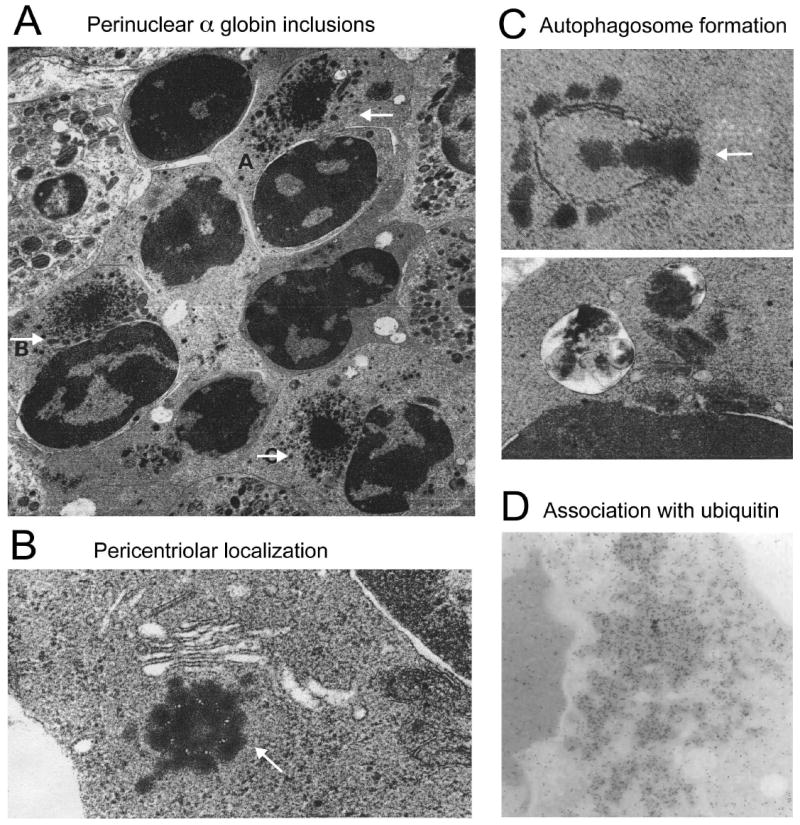
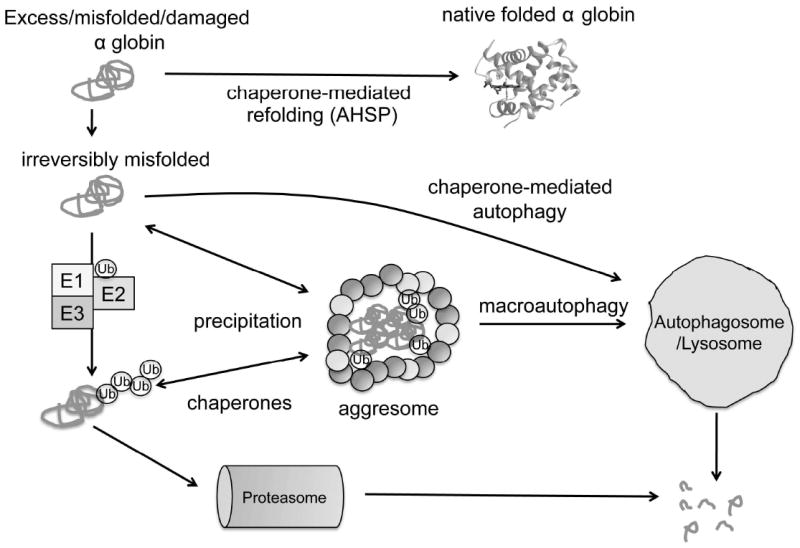
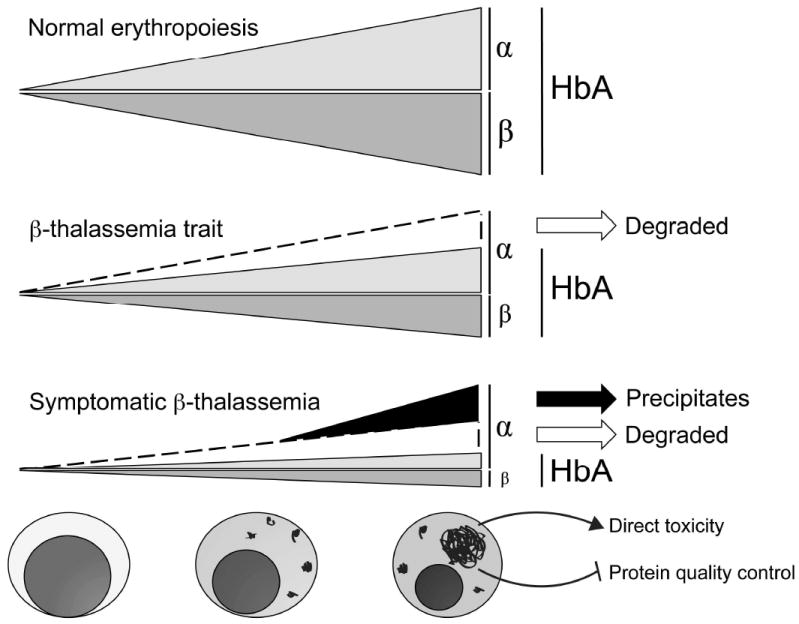
Erythrocytes must regulate hemoglobin synthesis to limit the toxicities of unstable free globin chain subunits. This regulation is particularly relevant in β-thalassemia, in which β-globin deficiency causes accumulation of free α-globin, which forms intracellular precipitates that destroy erythroid precursors. Experimental evidence accumulated over more than 40 years indicates that erythroid cells can neutralize moderate amounts of free α-globin through generalized protein quality control mechanisms, including molecular chaperones, the ubiquitin-proteasome system, and autophagy. In many ways, β-thalassemia resembles protein aggregation disorders of the nervous system, liver, and other tissues, which occur when levels of unstable proteins overwhelm cellular compensatory mechanisms. Information gained from studies of nonerythroid protein aggregation disorders may be exploited to further understand and perhaps treat β-thalassemia.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia.Blood. 2012 May 31;119(22):5265-75. doi: 10.1182/blood-2011-12-397729. Epub 2012 Mar 16. Blood. 2012. PMID: 22427201 Free PMC article. Clinical Trial.
-
Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus.Int J Mol Sci. 2023 Oct 10;24(20):15049. doi: 10.3390/ijms242015049. Int J Mol Sci. 2023. PMID: 37894732 Free PMC article.
-
Alpha-hemoglobin stabilizing protein: molecular function and clinical correlation.Front Biosci (Landmark Ed). 2010 Jan 1;15(1):1-11. doi: 10.2741/3601. Front Biosci (Landmark Ed). 2010. PMID: 20036801 Review.
-
Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia.J Clin Invest. 2004 Nov;114(10):1457-66. doi: 10.1172/JCI21982. J Clin Invest. 2004. PMID: 15545996 Free PMC article.
-
The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia.Int J Mol Sci. 2022 Sep 16;23(18):10811. doi: 10.3390/ijms231810811. Int J Mol Sci. 2022. PMID: 36142738 Free PMC article. Review.
Cited by
-
Discovery-Based Proteomics Identify Skeletal Muscle Mitochondrial Alterations as an Early Metabolic Defect in a Mouse Model of β-Thalassemia.Int J Mol Sci. 2023 Feb 23;24(5):4402. doi: 10.3390/ijms24054402. Int J Mol Sci. 2023. PMID: 36901833 Free PMC article.
-
Loss of miR-144/451 alleviates β-thalassemia by stimulating ULK1-mediated autophagy of free α-globin.Blood. 2023 Sep 7;142(10):918-932. doi: 10.1182/blood.2022017265. Blood. 2023. PMID: 37339583 Free PMC article.
-
Smad2/3-pathway ligand trap luspatercept enhances erythroid differentiation in murine β-thalassaemia by increasing GATA-1 availability.J Cell Mol Med. 2020 Jun;24(11):6162-6177. doi: 10.1111/jcmm.15243. Epub 2020 Apr 29. J Cell Mol Med. 2020. PMID: 32351032 Free PMC article.
-
Red chili powder dietary supplementation regularized the performance, hematobiochemical indices, oxidative status, and 8-hydroxy-2'-deoxyguanosine of aflatoxin B1 exposed broiler chickens.Transl Anim Sci. 2024 Jan 11;8:txae006. doi: 10.1093/tas/txae006. eCollection 2024. Transl Anim Sci. 2024. PMID: 38313223 Free PMC article.
-
Human cellular model systems of β-thalassemia enable in-depth analysis of disease phenotype.Nat Commun. 2023 Oct 6;14(1):6260. doi: 10.1038/s41467-023-41961-9. Nat Commun. 2023. PMID: 37803026 Free PMC article.
References
-
- Weatherall DJ, Clegg JB. The thalassaemia syndromes. 4. Oxford ; Malden, MA: Blackwell Science; 2001.
-
- Aigelsreiter A, Janig E, Stumptner C, et al. How a cell deals with abnormal proteins. Pathogenetic mechanisms in protein aggregation diseases. Pathobiology. 2007;74(3):145–158. - PubMed
-
- Garcia-Mata R, Gao Y-S, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002;3(6):388–396. - PubMed
-
- Ding W-X, Yin X-M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4(2):141–150. - PubMed
-
- Angastiniotis M, Modell B. Global epidemiology of hemoglobin disorders. Ann N Y Acad Sci. 1998;850:251–269. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
