

Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in β-thalassemia
- PMID: 21075282
- PMCID: PMC2991049
- DOI: 10.1016/j.hoc.2010.08.003
Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in β-thalassemia
Abstract
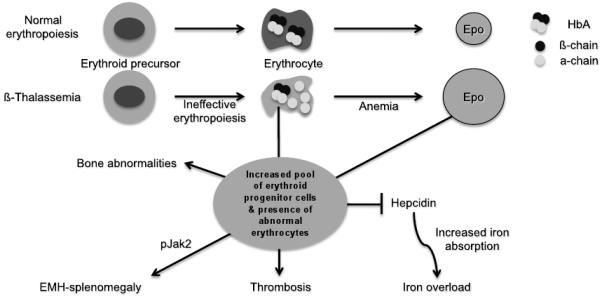
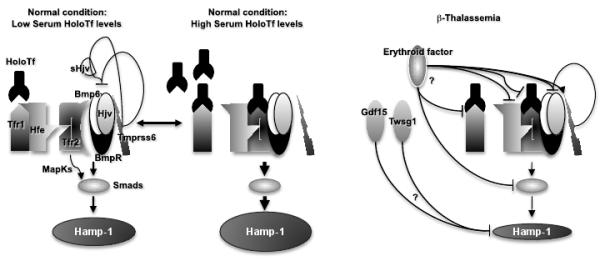
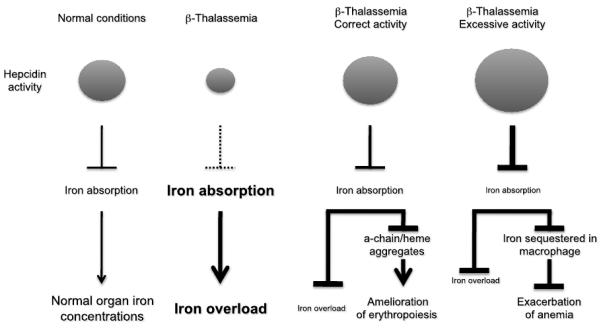
β-Thalassemia is a genetic disorder caused by mutations in the β-globin gene and characterized by chronic anemia caused by ineffective erythropoiesis, and accompanied by a variety of serious secondary complications such as extramedullary hematopoiesis, splenomegaly, and iron overload. In the past few years, numerous studies have shown that such secondary disease conditions have a genetic basis caused by the abnormal expression of genes with a role in controlling erythropoiesis and iron metabolism. In this article, the most recent discoveries related to the mechanism(s) responsible for anemia/ineffective erythropoiesis and iron overload are discussed in detail. Particular attention is paid to the pathway(s) controlling the expression of hepcidin, which is the main regulator of iron metabolism, and the Epo/EpoR/Jak2/Stat5 signaling pathway, which regulates erythropoiesis. Better understanding of how these pathways function and are altered in β-thalassemia has revealed several possibilities for development of new therapeutic approaches to treat of the complications of this disease.
Published by Elsevier Inc.
Figures
References
-
- Cooley TB, Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans. Am. Pediatr. Soc. 1925;37:29.
-
- Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2(4):245–55. - PubMed
-
- Forget BG. Molecular mechanisms of ß thalassemia. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. Cambridge University Press; Cambridge, UK: 2001. pp. 252–276.
-
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. 2nd ed. Cambridge University Press; Cambridge, UK: 2009. pp. 1–826.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
