

A stapled p53 helix overcomes HDMX-mediated suppression of p53
- PMID: 21075307
- PMCID: PMC3050021
- DOI: 10.1016/j.ccr.2010.10.024
A stapled p53 helix overcomes HDMX-mediated suppression of p53
Abstract
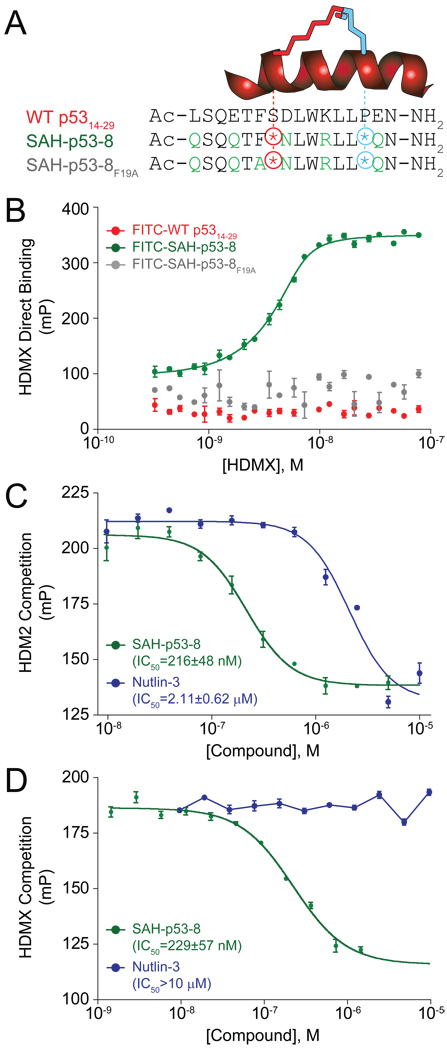
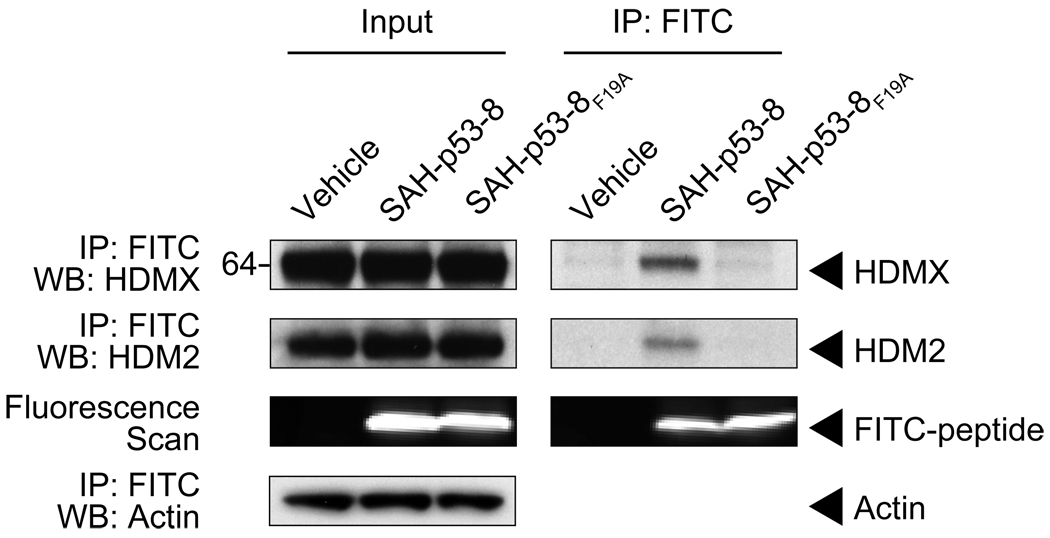
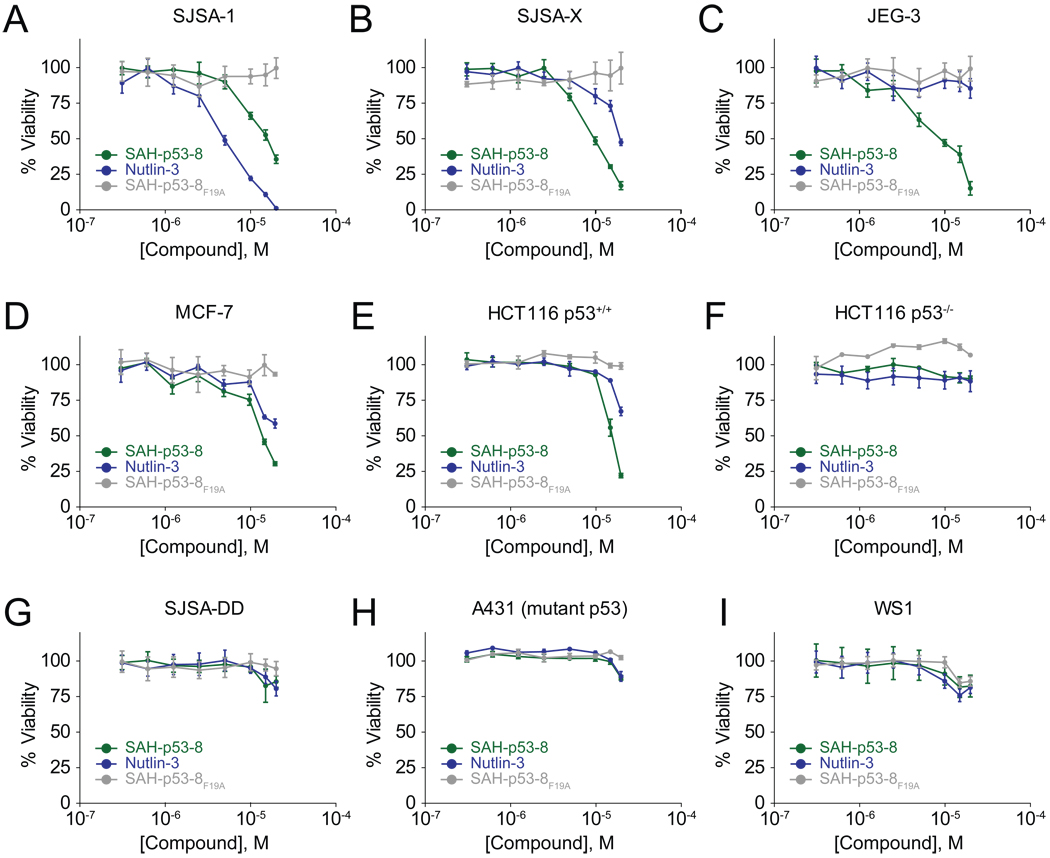
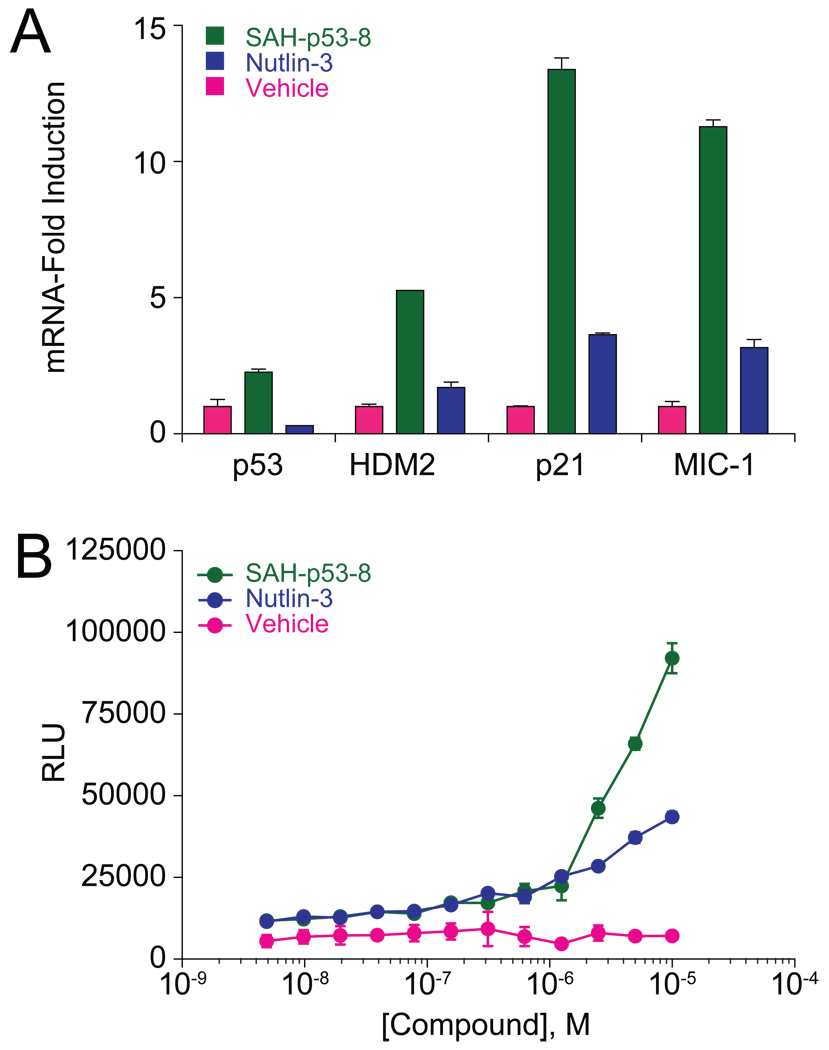
Cancer cells neutralize p53 by deletion, mutation, proteasomal degradation, or sequestration to achieve a pathologic survival advantage. Targeting the E3 ubiquitin ligase HDM2 can lead to a therapeutic surge in p53 levels. However, the efficacy of HDM2 inhibition can be compromised by overexpression of HDMX, an HDM2 homolog that binds and sequesters p53. Here, we report that a stapled p53 helix preferentially targets HDMX, blocks the formation of inhibitory p53-HDMX complexes, induces p53-dependent transcriptional upregulation, and thereby overcomes HDMX-mediated cancer resistance in vitro and in vivo. Importantly, our analysis of p53 interaction dynamics provides a blueprint for reactivating the p53 pathway in cancer by matching HDM2, HDMX, or dual inhibitors to the appropriate cellular context.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures

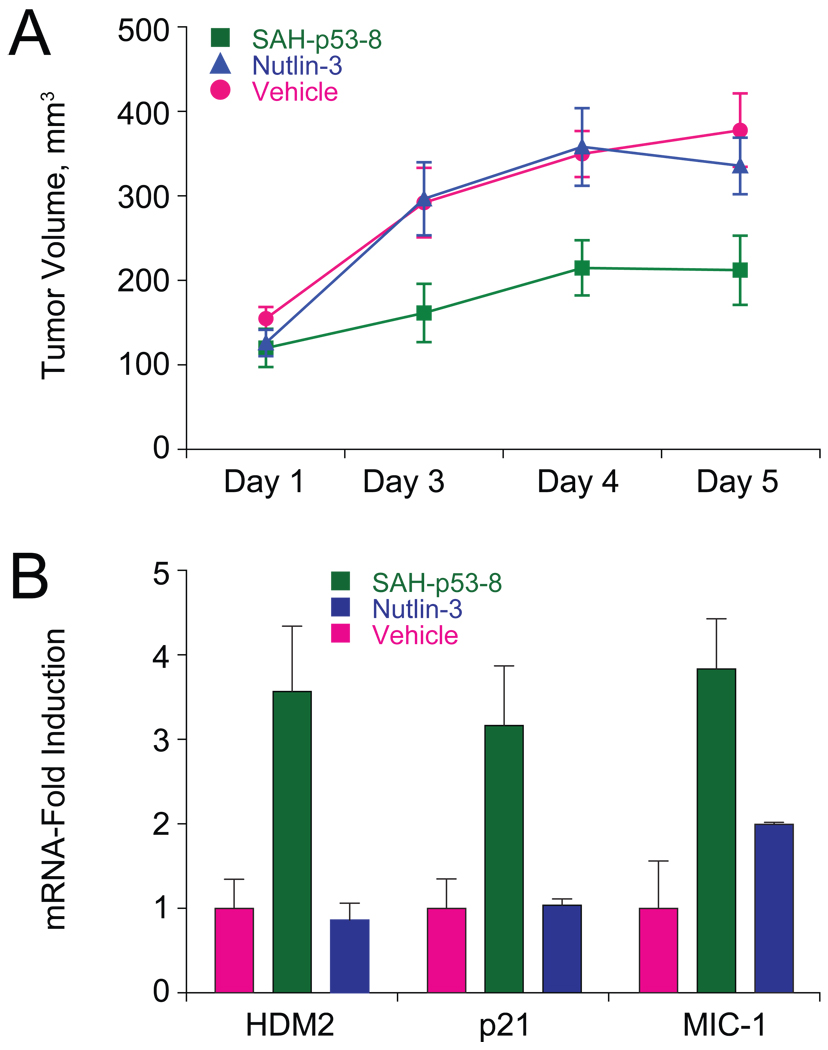
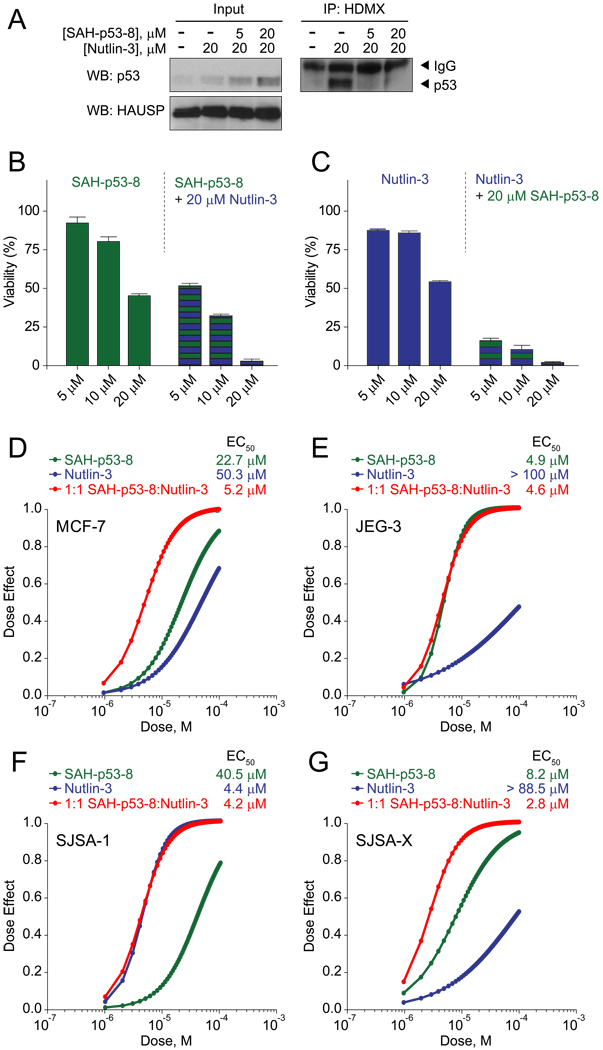

Comment on
-
Pharmacological rescue of p53 in cancer therapy: widening the sensitive tumor spectrum by targeting MDMX.Cancer Cell. 2010 Nov 16;18(5):399-400. doi: 10.1016/j.ccr.2010.10.026. Cancer Cell. 2010. PMID: 21075301 No abstract available.
References
-
- Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. - PubMed
-
- Böttger A, Böttger V, Garcia-Echeverria C, Chène P, Hochkeppel HK, Sampson W, Ang K, Howard SF, Picksley SM, Lane DP. Molecular characterization of the hdm2-p53 interaction. J Mol Biol. 1997;269:744–756. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
