

Efficient parallel and out of core algorithms for constructing large bi-directed de Bruijn graphs
- PMID: 21078174
- PMCID: PMC2996408
- DOI: 10.1186/1471-2105-11-560
Efficient parallel and out of core algorithms for constructing large bi-directed de Bruijn graphs
Abstract
Background: Assembling genomic sequences from a set of overlapping reads is one of the most fundamental problems in computational biology. Algorithms addressing the assembly problem fall into two broad categories - based on the data structures which they employ. The first class uses an overlap/string graph and the second type uses a de Bruijn graph. However with the recent advances in short read sequencing technology, de Bruijn graph based algorithms seem to play a vital role in practice. Efficient algorithms for building these massive de Bruijn graphs are very essential in large sequencing projects based on short reads. In an earlier work, an O(n/p) time parallel algorithm has been given for this problem. Here n is the size of the input and p is the number of processors. This algorithm enumerates all possible bi-directed edges which can overlap with a node and ends up generating Θ(nΣ) messages (Σ being the size of the alphabet).
Results: In this paper we present a Θ(n/p) time parallel algorithm with a communication complexity that is equal to that of parallel sorting and is not sensitive to Σ. The generality of our algorithm makes it very easy to extend it even to the out-of-core model and in this case it has an optimal I/O complexity of Θ(nlog(n/B)Blog(M/B)) (M being the main memory size and B being the size of the disk block). We demonstrate the scalability of our parallel algorithm on a SGI/Altix computer. A comparison of our algorithm with the previous approaches reveals that our algorithm is faster--both asymptotically and practically. We demonstrate the scalability of our sequential out-of-core algorithm by comparing it with the algorithm used by VELVET to build the bi-directed de Bruijn graph. Our experiments reveal that our algorithm can build the graph with a constant amount of memory, which clearly outperforms VELVET. We also provide efficient algorithms for the bi-directed chain compaction problem.
Conclusions: The bi-directed de Bruijn graph is a fundamental data structure for any sequence assembly program based on Eulerian approach. Our algorithms for constructing Bi-directed de Bruijn graphs are efficient in parallel and out of core settings. These algorithms can be used in building large scale bi-directed de Bruijn graphs. Furthermore, our algorithms do not employ any all-to-all communications in a parallel setting and perform better than the prior algorithms. Finally our out-of-core algorithm is extremely memory efficient and can replace the existing graph construction algorithm in VELVET.
Figures
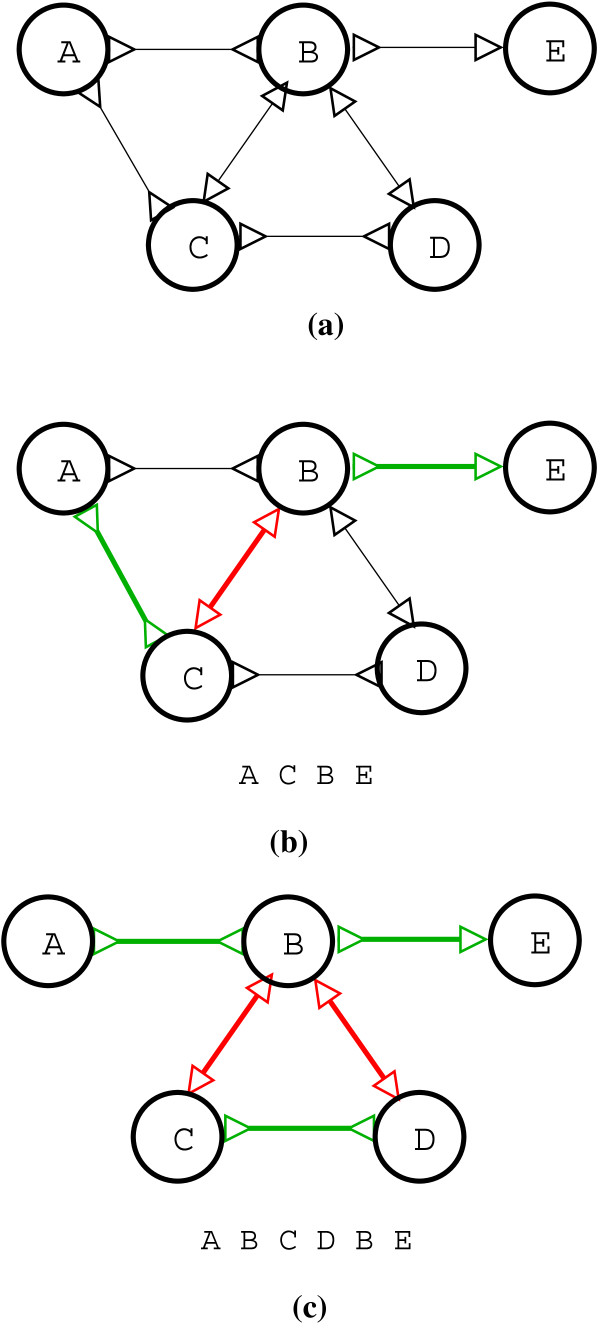
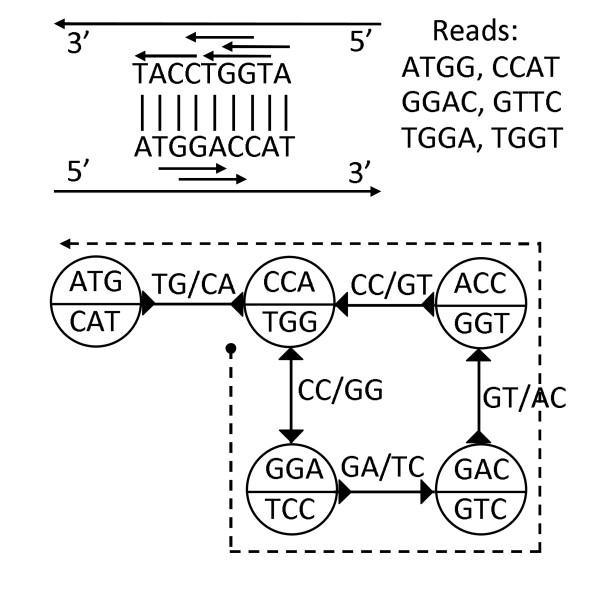
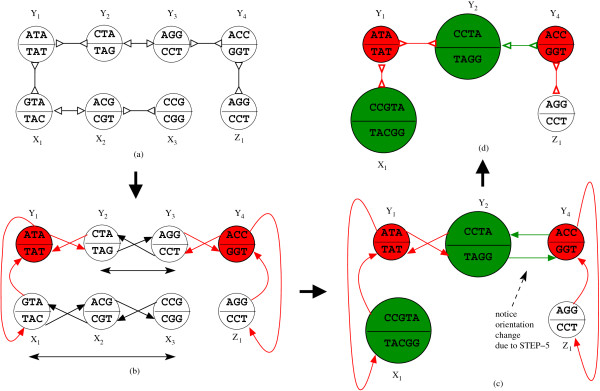
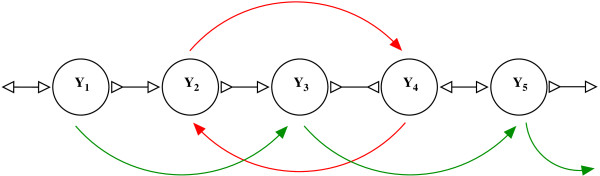
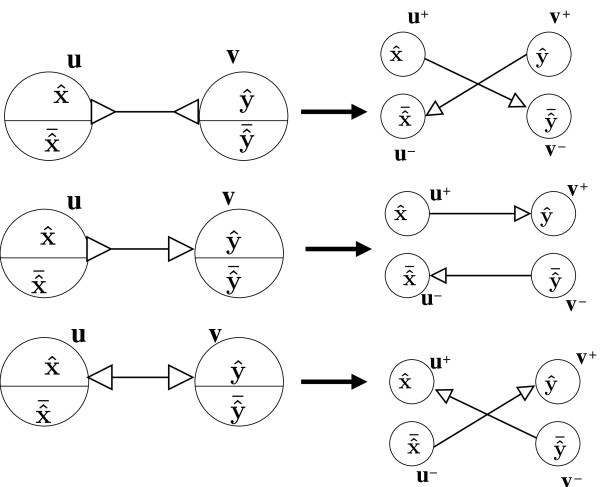
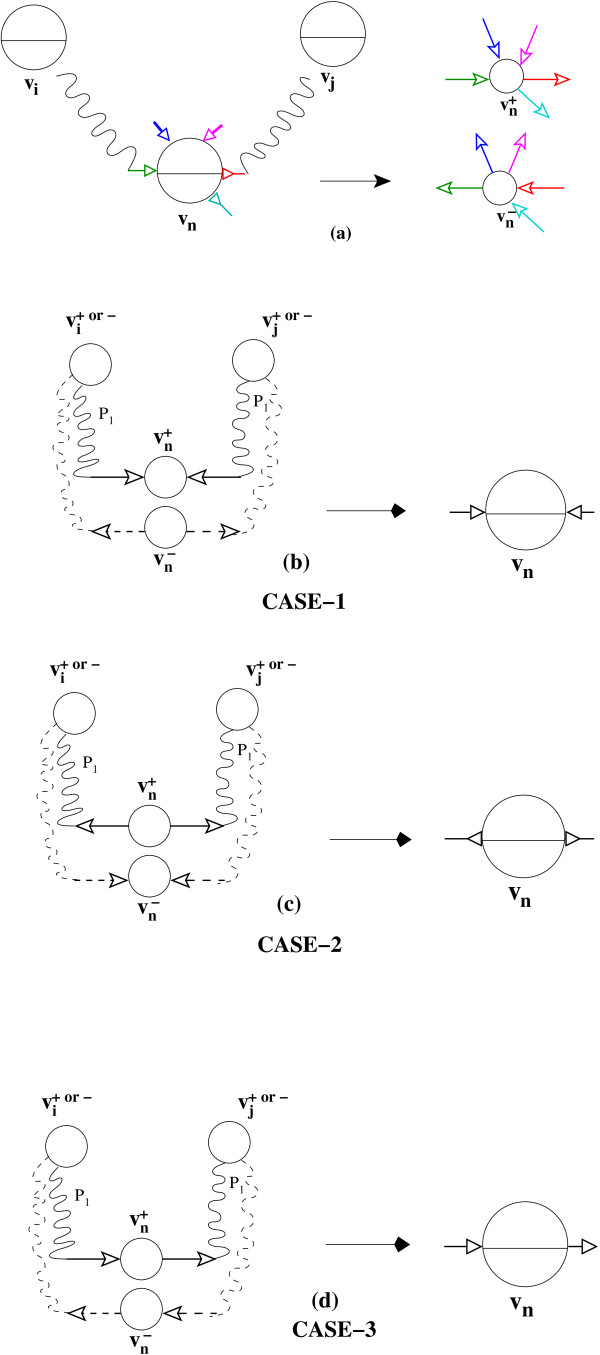
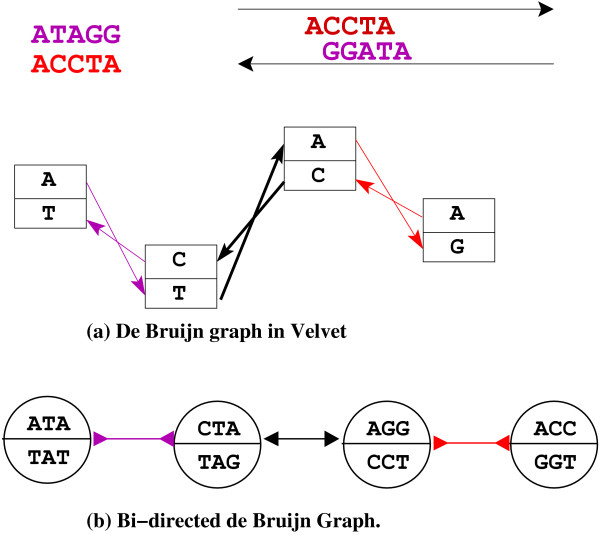
Similar articles
-
AN EFFICIENT ALGORITHM FOR CHINESE POSTMAN WALK ON BI-DIRECTED DE BRUIJN GRAPHS.Discrete Math Algorithms Appl. 2010;1:184-196. doi: 10.1007/978-3-642-17458-2_16. Discrete Math Algorithms Appl. 2010. PMID: 25364472 Free PMC article.
-
Fast de Bruijn Graph Compaction in Distributed Memory Environments.IEEE/ACM Trans Comput Biol Bioinform. 2020 Jan-Feb;17(1):136-148. doi: 10.1109/TCBB.2018.2858797. Epub 2018 Jul 31. IEEE/ACM Trans Comput Biol Bioinform. 2020. PMID: 30072337
-
On the Hardness of Sequence Alignment on De Bruijn Graphs.J Comput Biol. 2022 Dec;29(12):1377-1396. doi: 10.1089/cmb.2022.0411. Epub 2022 Nov 25. J Comput Biol. 2022. PMID: 36450127
-
Applications of de Bruijn graphs in microbiome research.Imeta. 2022 Mar 1;1(1):e4. doi: 10.1002/imt2.4. eCollection 2022 Mar. Imeta. 2022. PMID: 38867733 Free PMC article. Review.
-
The present and future of de novo whole-genome assembly.Brief Bioinform. 2018 Jan 1;19(1):23-40. doi: 10.1093/bib/bbw096. Brief Bioinform. 2018. PMID: 27742661 Review.
Cited by
-
Cuttlefish: fast, parallel and low-memory compaction of de Bruijn graphs from large-scale genome collections.Bioinformatics. 2021 Jul 12;37(Suppl_1):i177-i186. doi: 10.1093/bioinformatics/btab309. Bioinformatics. 2021. PMID: 34252958 Free PMC article.
-
Breaking the computational barriers of pairwise genome comparison.BMC Bioinformatics. 2015 Aug 11;16(1):250. doi: 10.1186/s12859-015-0679-9. BMC Bioinformatics. 2015. PMID: 26260162 Free PMC article.
-
Compacting de Bruijn graphs from sequencing data quickly and in low memory.Bioinformatics. 2016 Jun 15;32(12):i201-i208. doi: 10.1093/bioinformatics/btw279. Bioinformatics. 2016. PMID: 27307618 Free PMC article.
-
Exploring variation-aware contig graphs for (comparative) metagenomics using MaryGold.Bioinformatics. 2013 Nov 15;29(22):2826-34. doi: 10.1093/bioinformatics/btt502. Epub 2013 Sep 20. Bioinformatics. 2013. PMID: 24058058 Free PMC article.
References
-
- Kececioglu JD, Myers EW. Combinatorial algorithms for DNA sequence assembly. Algorithmica. 1995;13(1-2):7–51. doi: 10.1007/BF01188580. - DOI
-
- Medvedev P, Georgiou K, Myers G, Brudno M. Computability of models for sequence assembly. Workshop on Algorithms for Bioinformatics (WABI), LNBI-4645. 2007. pp. 289–301. full_text.
-
- Huang X, Wang J, Aluru S, Yang S, Hillier L. PCAP: A whole-genome assembly program. Genome research. 2003;13(9):2164–2170. doi: 10.1101/gr.1390403. http://www.scopus.com [Cited By (since 1996): 61] - DOI - PMC - PubMed
-
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KHJ, Remington KA, Anson EL, Bolanos RA, Chou H, Jordan CM, Halpern AL, Lonardi S, Beasley EM, Brandon RC, Chen L, Dunn PJ, Lai Z, Liang Y, Nusskern DR, Zhan M, Zhang Q, Zheng X, Rubin GM, Adams MD, Venter JC. A whole-genome assembly of Drosophila. Science. 2000;287(5461):2196–2204. doi: 10.1126/science.287.5461.2196. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
