

Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production
- PMID: 21079150
- PMCID: PMC3056471
- DOI: 10.1182/blood-2010-05-287821
Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production
Abstract
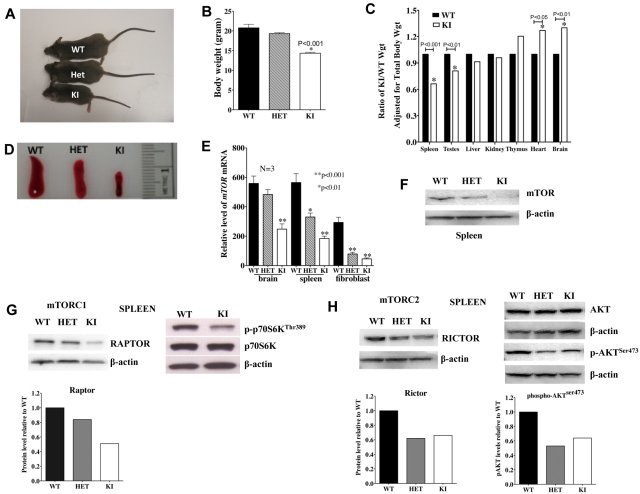
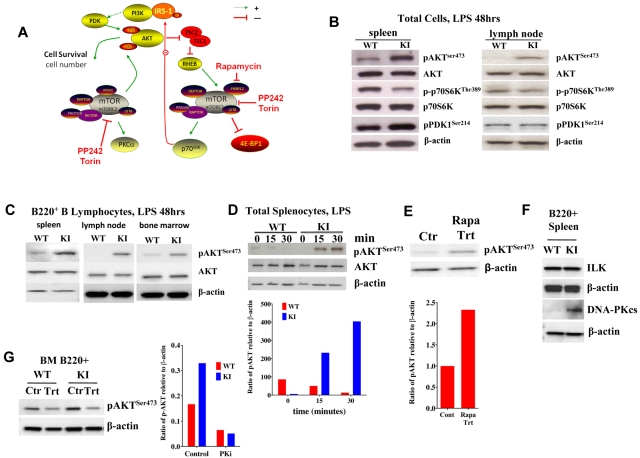
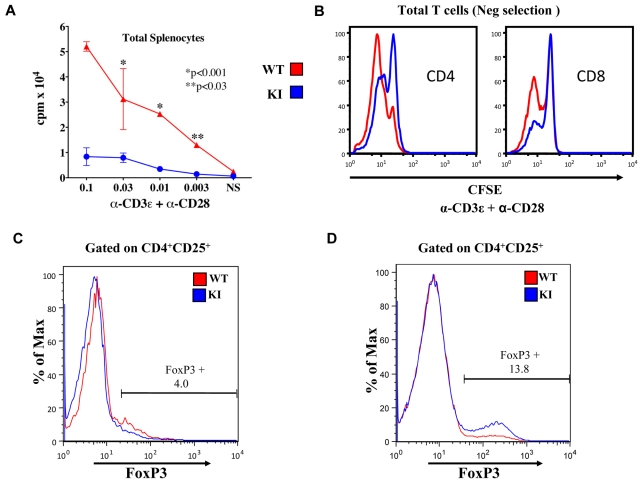
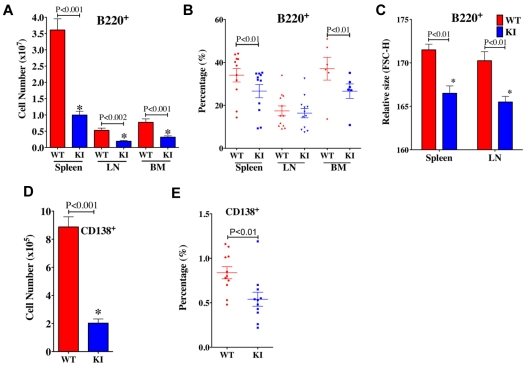
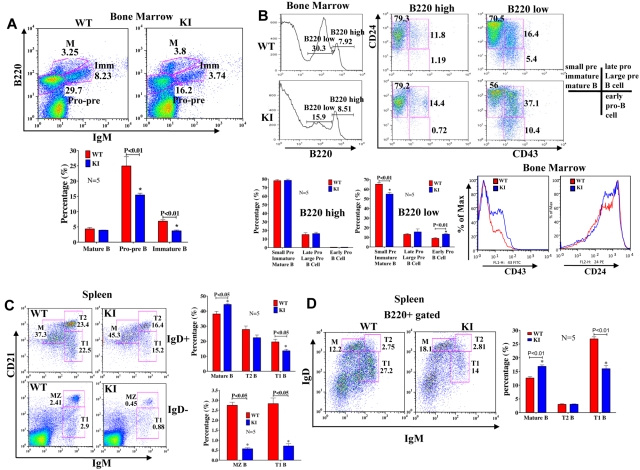
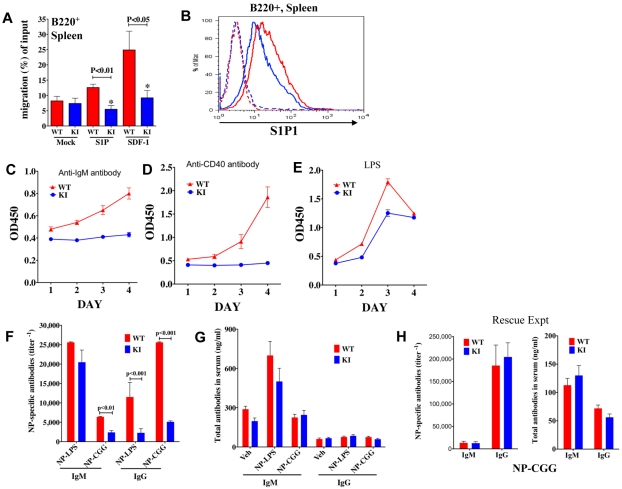
Mammalian TOR (mTOR) regulates cell growth, proliferation, and migration. Because mTOR knock-outs are embryonic lethal, we generated a viable hypomorphic mouse by neo-insertion that partially disrupts mTOR transcription and creates a potential physiologic model of mTORC1/TORC2 inhibition. Homozygous knock-in mice exhibited reductions in body, organ, and cell size. Although reductions in most organ sizes were proportional to decreased body weight, spleens were disproportionately smaller. Decreases in the total number of T cells, particularly memory cells, and reduced responses to chemokines suggested alterations in T-cell homing/homeostasis. T-cell receptor-stimulated T cells proliferated less, produced lower cytokine levels, and expressed FoxP3. Decreased neutrophil numbers were also observed in the spleen, despite normal development and migration in the bone marrow. However, B-cell effects were most pronounced, with a partial block in B-cell development in the bone marrow, altered splenic populations, and decreases in proliferation, antibody production, and migration to chemokines. Moreover, increased AKT(Ser473) phosphorylation was observed in activated B cells, reminiscent of cancers treated with rapamycin, and was reduced by a DNA-pk inhibitor. Thus, mTOR is required for the maturation and differentiation of multiple immune cell lineages. These mice provide a novel platform for studying the consequences of constitutively reduced mTORC1/TORC2 activity.
Figures
References
-
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. - PubMed
-
- Yang Q, Guan KL. Expanding mTOR signaling. Cell Res. 2007;17(8):666–681. - PubMed
-
- Astrinidis A, Cash TP, Hunter DS, Walker CL, Chernoff J, Henske EP. Tuberin, the tuberous sclerosis complex 2 tumor suppressor gene product, regulates Rho activation, cell adhesion and migration. Oncogene. 2002;21(55):8470–8476. - PubMed
-
- Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
