

Review
doi: 10.1083/jcb.201006173.
The cell biology of polycystic kidney disease
Affiliations
- PMID: 21079243
- PMCID: PMC2983067
- DOI: 10.1083/jcb.201006173
Item in Clipboard
Review
The cell biology of polycystic kidney disease
J Cell Biol.
.
Abstract
Polycystic kidney disease is a common genetic disorder in which fluid-filled cysts displace normal renal tubules. Here we focus on autosomal dominant polycystic kidney disease, which is attributable to mutations in the PKD1 and PKD2 genes and which is characterized by perturbations of renal epithelial cell growth control, fluid transport, and morphogenesis. The mechanisms that connect the underlying genetic defects to disease pathogenesis are poorly understood, but their exploration is shedding new light on interesting cell biological processes and suggesting novel therapeutic targets.
Figures

N- and C-terminal cleavage of the PC1 protein. The N terminus of PC1 is cleaved at the G protein–coupled receptor proteolytic site (GPS), but the extracellular domain remains noncovalently attached to the membrane-bound portion of the protein. Either of two different cleavages can release C-terminal tail fragments that translocate to the nucleus with components of the Wnt pathway, STAT6/p100, and perhaps with other regulators of transcription. At least one of the C-terminal tail cleavages is stimulated by the presence of PC2, and this stimulation requires that PC2 be capable of functioning as an ion channel.
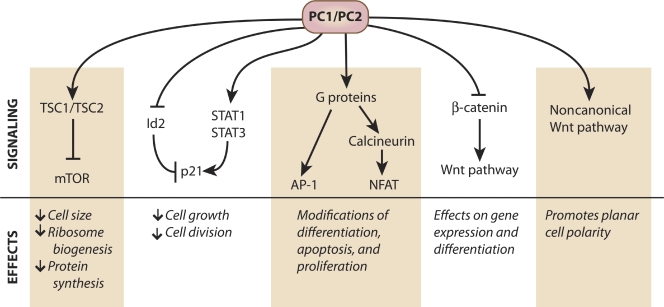
PC1 and PC2 affect multiple signaling pathways. Summary of the effects that PC1 and PC2 exert on signaling pathways. Multiple direct and indirect interactions allow the polycystin proteins to inhibit or stimulate pathways involved in cellular growth and differentiation.

Cyst formation at the level of the cell, nephron, and kidney. Defects in the genes encoding PC1 or PC2 lead to aberrant gene transcription, cell proliferation, and ion secretion, which in turn result in the formation of fluid-filled cysts. As cysts balloon out from individual nephrons, their collective effect leads to the displacement of the normal renal parenchyma and the formation of a cyst-filled kidney with reduced functional capacity.
References
-
- Arnould T., Kim E., Tsiokas L., Jochimsen F., Grüning W., Chang J.D., Walz G. 1998. The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J. Biol. Chem. 273:6013–6018 10.1074/jbc.273.11.6013 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
