

A reappraisal of Gaucher disease-diagnosis and disease management algorithms
- PMID: 21080341
- PMCID: PMC3058841
- DOI: 10.1002/ajh.21888
A reappraisal of Gaucher disease-diagnosis and disease management algorithms
Abstract
Type 1 (non-neuronopathic) Gaucher disease was the first lysosomal storage disorder for which an effective enzyme replacement therapy was developed and it has become a prototype for treatments for related orphan diseases. There are currently four treatment options available to patients with Gaucher disease, nevertheless, almost 25% of Type 1 Gaucher patients do not gain timely access to therapy because of delays in diagnosis after the onset of symptoms. Diagnosis of Gaucher disease by enzyme testing is unequivocal, but the rarity of the disease and nonspecific and heterogeneous nature of Gaucher disease symptoms may impede consideration of this disease in the differential diagnosis. To help promote timely diagnosis and optimal management of the protean presentations of Gaucher disease, a consensus meeting was convened to develop algorithms for diagnosis and disease management for Gaucher disease.
Figures
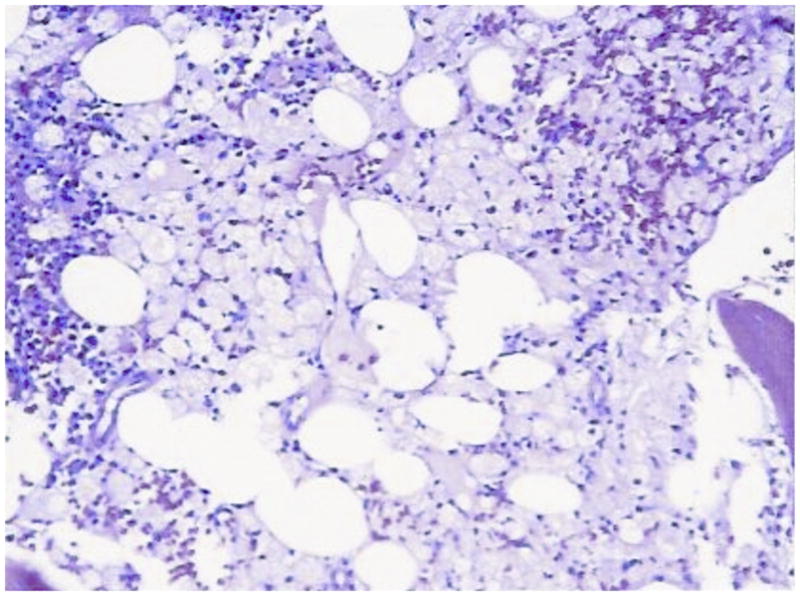
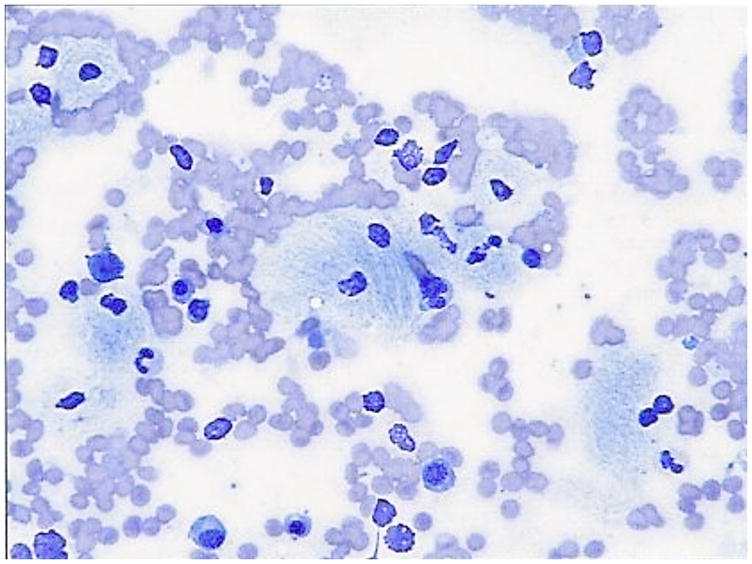
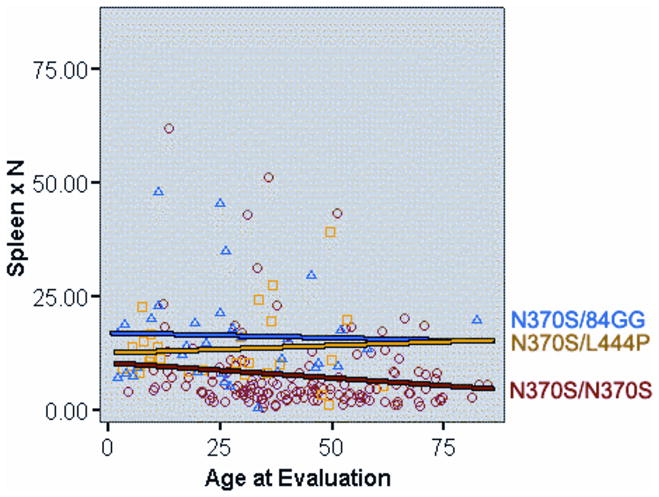
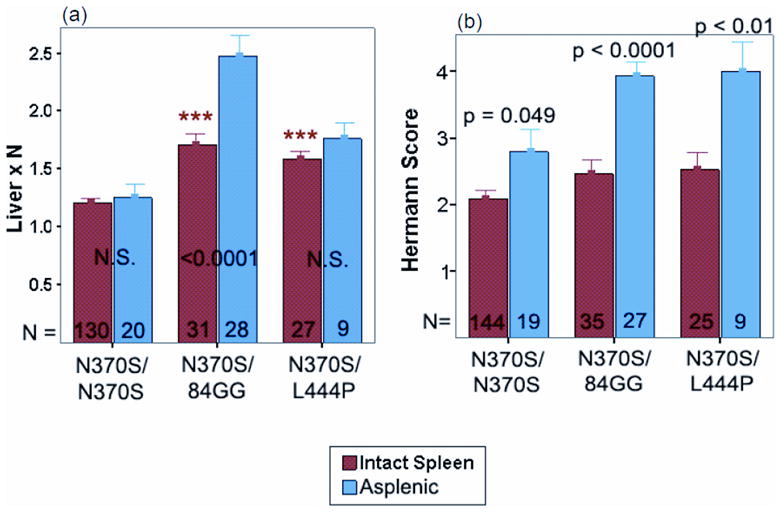

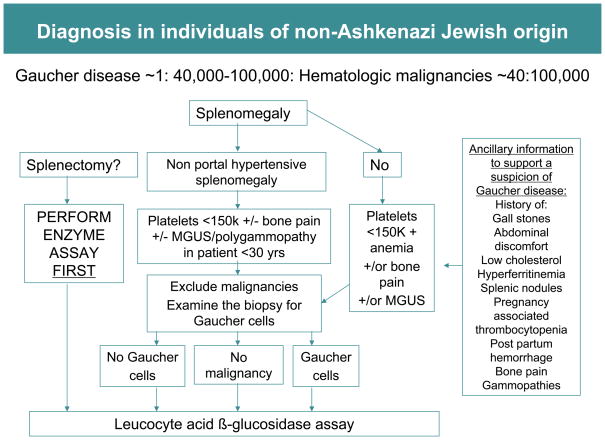
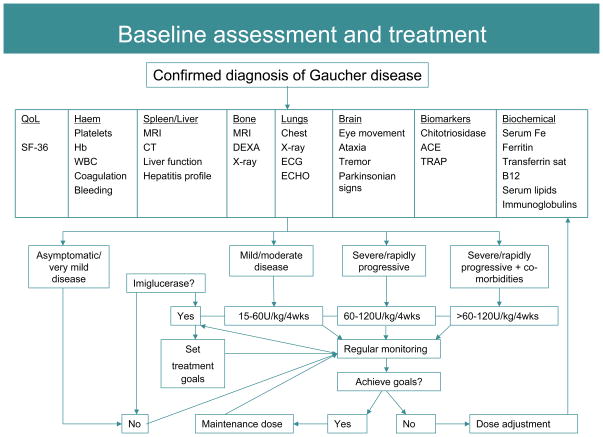
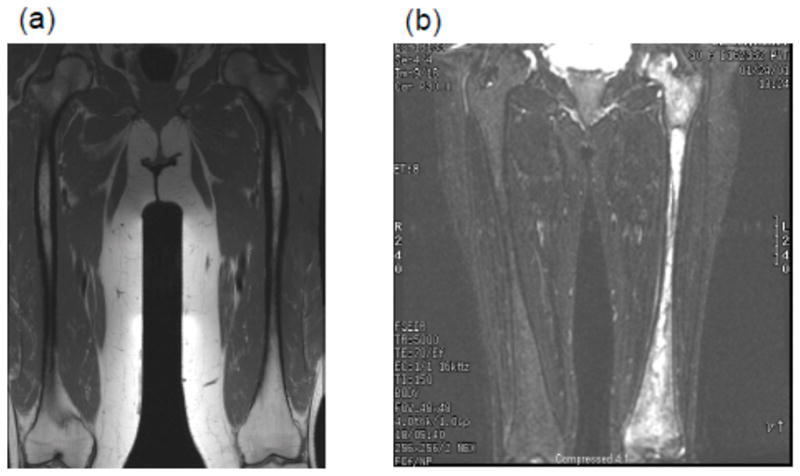
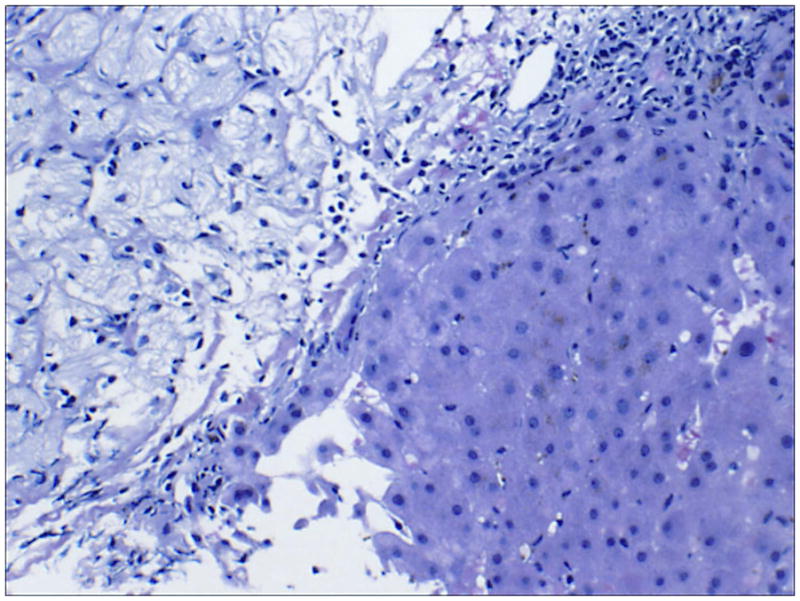
References
-
- Beutler E, Grabowski GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. New York, NY: McGraw-Hill; 2001. pp. 3635–3668.
-
- Zimran A, Altarescu G, Rudensky B, et al. Survey of hematological aspects of Gaucher disease. Hematology. 2005;10:151–156. - PubMed
-
- Cox TM, Schofield JP. Gaucher’s disease: clinical features and natural history. Baillieres Clin Haematol. 1997;10:657–89. - PubMed
-
- Grabowski GA. Gaucher disease: gene frequencies and genotype/phenotype correlations. Genet Test. 1997;1:5–12. - PubMed
-
- Horowitz M, Pasmanik-Chor M, Borochowitz Z, et al. Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum Mutat. 1998;12:240–244. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases