

RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I
- PMID: 21084468
- PMCID: PMC3020501
- DOI: 10.1128/JVI.01635-10
RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I
Abstract
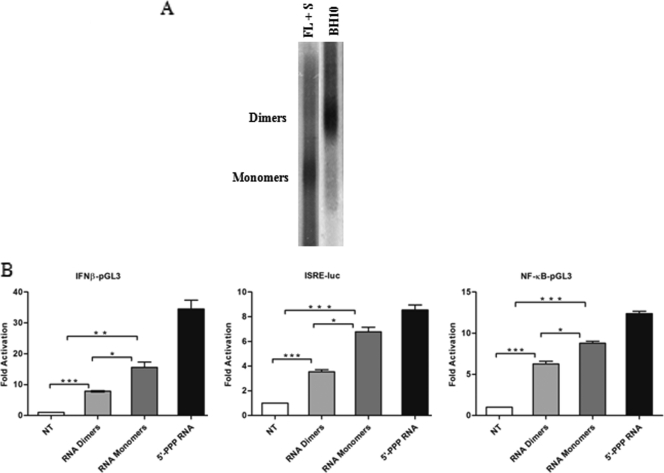
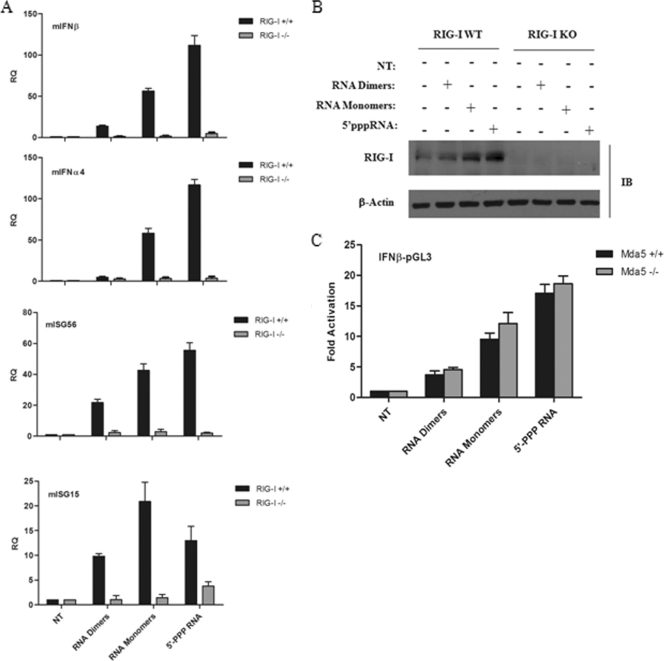
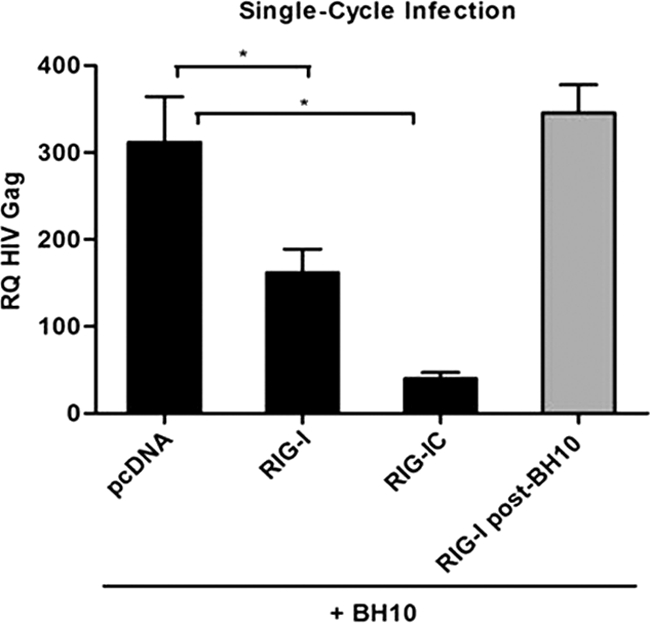
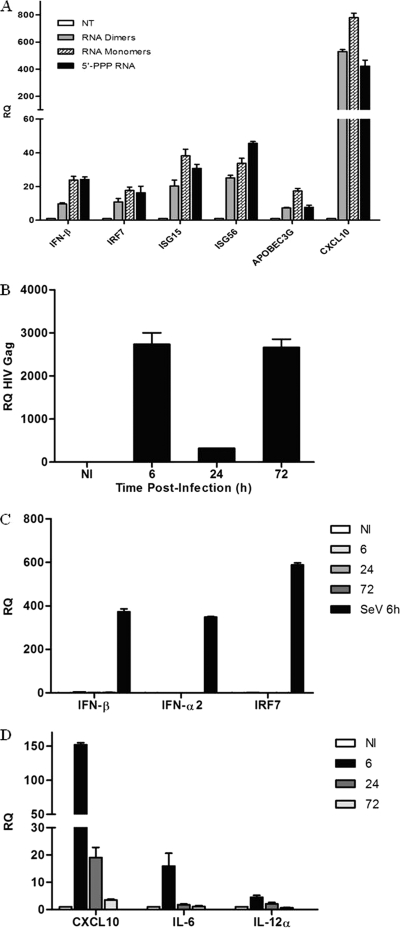
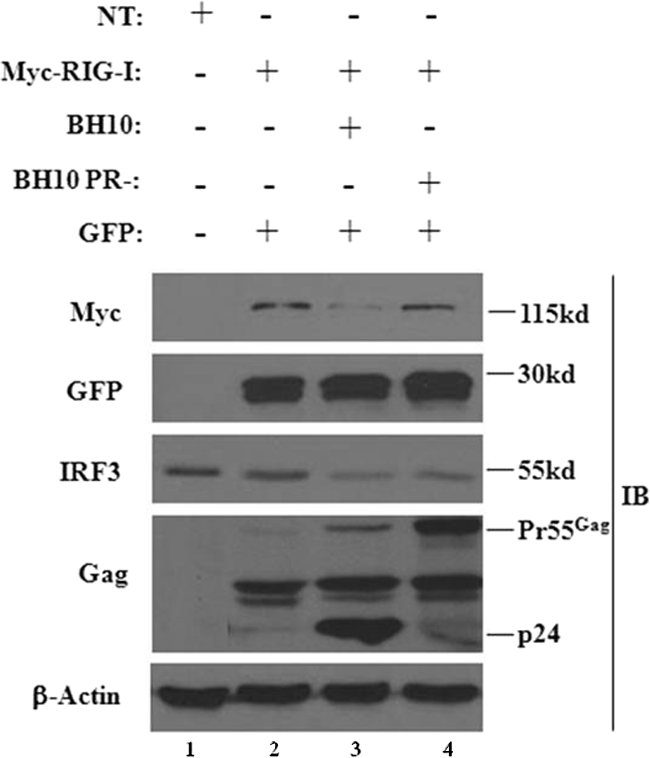
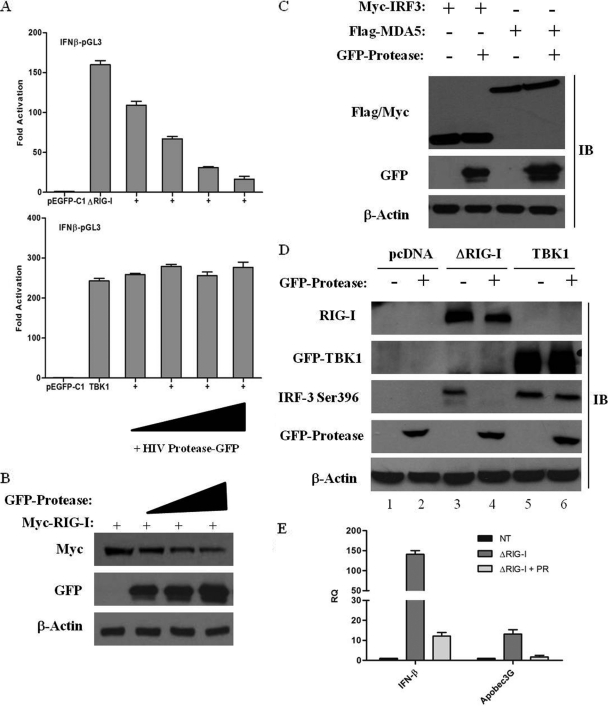
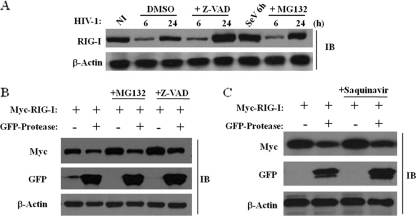
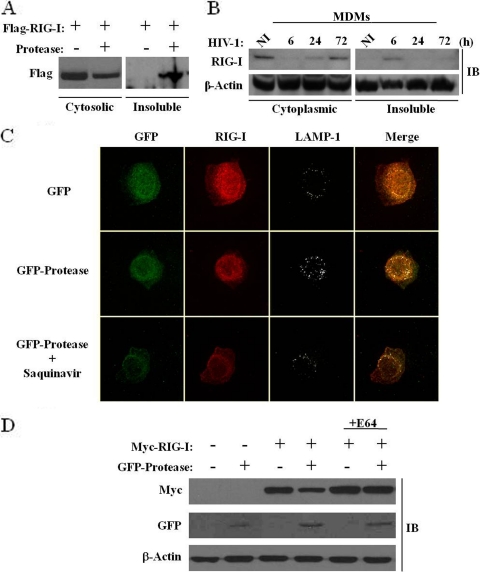
The rapid induction of type I interferon (IFN) is essential for establishing innate antiviral responses. During infection, cytoplasmic viral RNA is sensed by two DExD/H box RNA helicases, RIG-I and MDA5, ultimately driving IFN production. Here, we demonstrate that purified genomic RNA from HIV-1 induces a RIG-I-dependent type I IFN response. Both the dimeric and monomeric forms of HIV-1 were sensed by RIG-I, but not MDA5, with monomeric RNA, usually found in defective HIV-1 particles, acting as a better inducer of IFN than dimeric RNA. However, despite the presence of HIV-1 RNA in the de novo infection of monocyte-derived macrophages, HIV-1 replication did not lead to a substantial induction of IFN signaling. We demonstrate the existence of an evasion mechanism based on the inhibition of the RIG-I sensor through the action of the HIV-1 protease (PR). Indeed, the ectopic expression of PR resulted in the inhibition of IFN regulatory factor 3 (IRF-3) phosphorylation and decreased expression of IFN and interferon-stimulated genes. A downregulation of cytoplasmic RIG-I levels occurred in cells undergoing a single-cycle infection with wild-type provirus BH10 but not in cells transfected with a protease-deficient provirus, BH10-PR(-). Cellular fractionation and confocal microscopy studies revealed that RIG-I translocated from the cytosol to an insoluble fraction during the de novo HIV-1 infection of monocyte-derived macrophages, in the presence of PR. The loss of cytoplasmic RIG-I was prevented by the lysosomal inhibitor E64, suggesting that PR targets RIG-I to the lysosomes. This study reveals a novel PR-dependent mechanism employed by HIV-1 to counteract the early IFN response to viral RNA in infected cells.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
