

Genetic dissection of interferon-antagonistic functions of rabies virus phosphoprotein: inhibition of interferon regulatory factor 3 activation is important for pathogenicity
- PMID: 21084487
- PMCID: PMC3020028
- DOI: 10.1128/JVI.01427-10
Genetic dissection of interferon-antagonistic functions of rabies virus phosphoprotein: inhibition of interferon regulatory factor 3 activation is important for pathogenicity
Erratum in
- J Virol. 2012 Apr;86(8):4720
Abstract
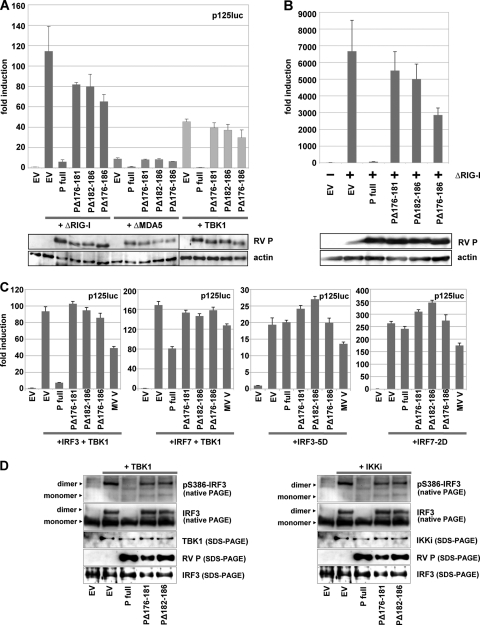
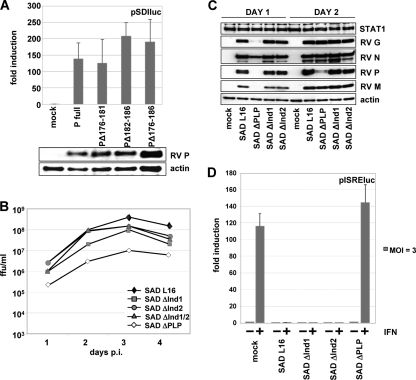
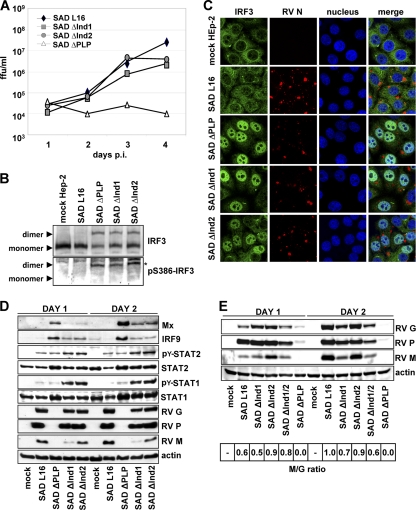
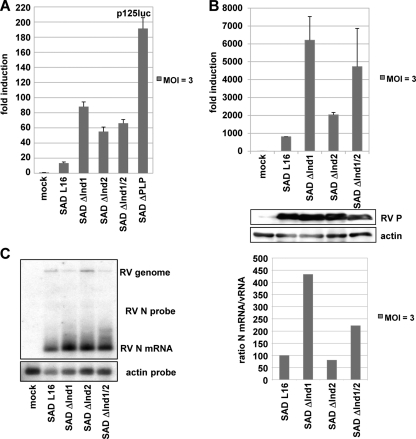
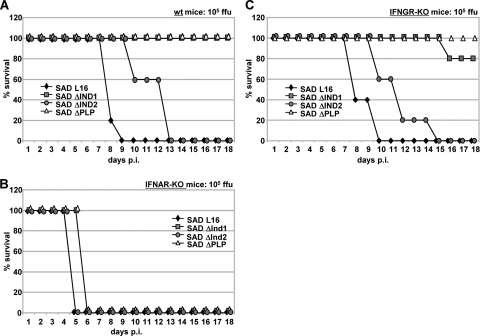
The rabies virus (RV) phosphoprotein (P) is a type I interferon (IFN) antagonist preventing both transcriptional induction of IFN and IFN-mediated JAK/STAT signaling. In addition, P is an essential cofactor of the viral polymerase and is required for encapsidation of viral RNA into nucleoprotein during replication. By site-directed mutagenesis, we have identified a domain of P required for efficient inhibition of IFN induction. Phosphoproteins lacking amino acids (aa) 176 to 181, 182 to 186, or 176 to 186 were severely compromised in counteracting phosphorylation of IRF3 and IRF7 by TBK1 or IKKi while retaining the full capacity of preventing nuclear import of activated STATs and of supporting virus transcription and replication. Recombinant RV carrying the mutated phosphoproteins (the SAD ΔInd1, SAD ΔInd2, and SAD ΔInd1/2 viruses) activated IRF3 and beta IFN (IFN-β) transcription in infected cells but still blocked STAT-mediated expression of IFN-stimulated genes. Due to a somewhat higher transcription rate, the SAD ΔInd1 virus activated IRF3 more efficiently than the SAD ΔInd2 virus. After intracerebral injection into mouse brains at high doses, the SAD ΔInd1 virus was completely apathogenic for wild-type (wt) mice, while the SAD ΔInd2 virus was partially attenuated and caused a slower progression of lethal rabies than wt RV. Neurovirulence of IFN-resistant RV thus correlates with the capacity of the virus to prevent activation of IRF3 and IRF7.
Figures
References
-
- Andersen, J., S. VanScoy, T. F. Cheng, D. Gomez, and N. C. Reich. 2008. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 9:168-175. - PubMed
-
- Blondel, D., et al. 2002. Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 21:7957-7970. - PubMed
-
- Brzózka, K., and K. K. Conzelmann. 2009. IFN escape of rhabdoviruses, p. 211-227. In A. Brasier and A. García-Sastre (ed.), Cellular signaling and innate immune responses to RNA virus infections. ASM Press, Washington, DC.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
