

Enrichment map: a network-based method for gene-set enrichment visualization and interpretation
- PMID: 21085593
- PMCID: PMC2981572
- DOI: 10.1371/journal.pone.0013984
Enrichment map: a network-based method for gene-set enrichment visualization and interpretation
Abstract
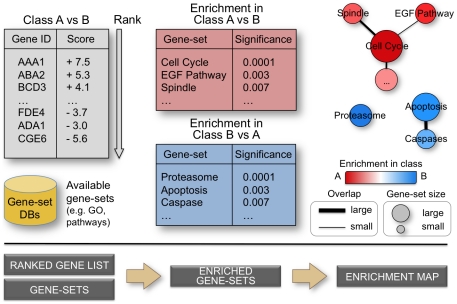
Background: Gene-set enrichment analysis is a useful technique to help functionally characterize large gene lists, such as the results of gene expression experiments. This technique finds functionally coherent gene-sets, such as pathways, that are statistically over-represented in a given gene list. Ideally, the number of resulting sets is smaller than the number of genes in the list, thus simplifying interpretation. However, the increasing number and redundancy of gene-sets used by many current enrichment analysis software works against this ideal.
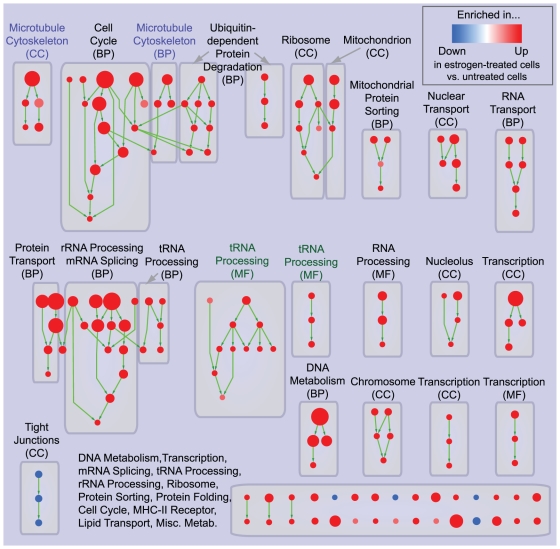
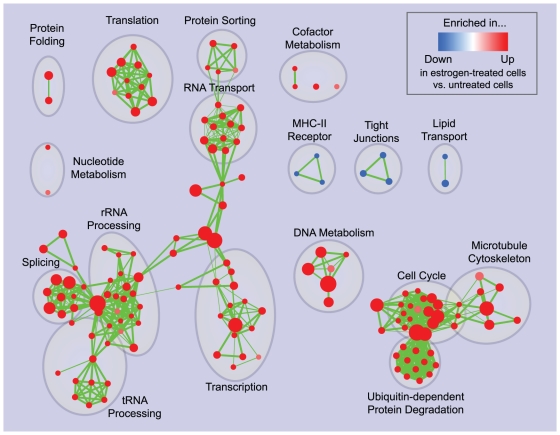
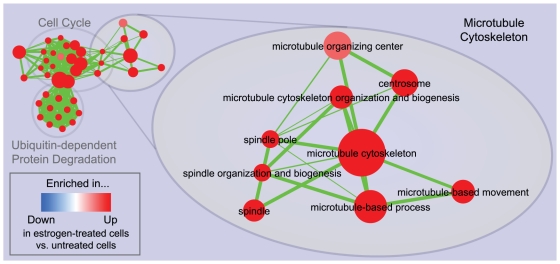
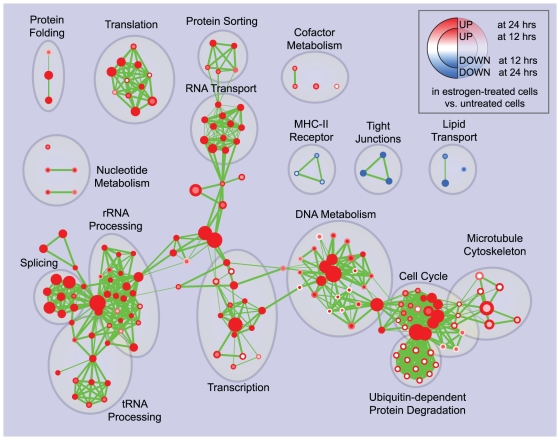
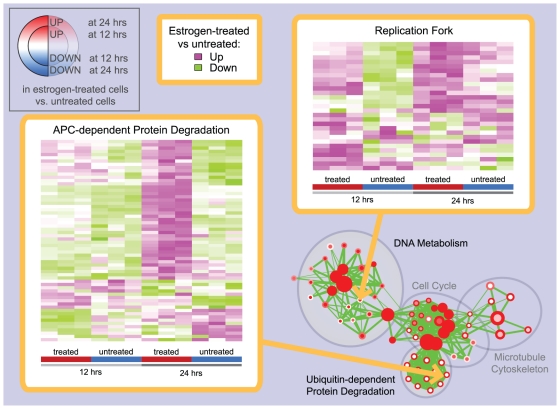
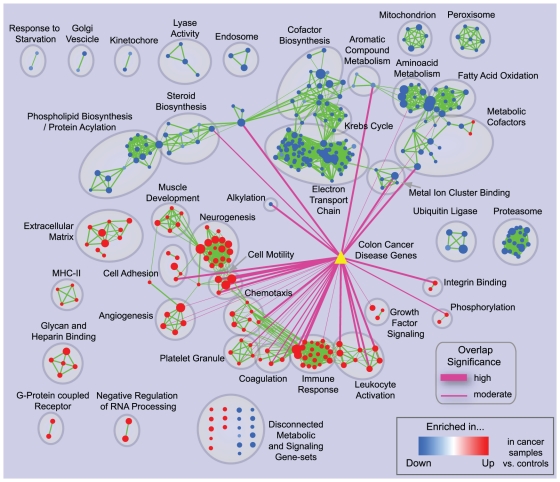
Principal findings: To overcome gene-set redundancy and help in the interpretation of large gene lists, we developed "Enrichment Map", a network-based visualization method for gene-set enrichment results. Gene-sets are organized in a network, where each set is a node and edges represent gene overlap between sets. Automated network layout groups related gene-sets into network clusters, enabling the user to quickly identify the major enriched functional themes and more easily interpret the enrichment results.
Conclusions: Enrichment Map is a significant advance in the interpretation of enrichment analysis. Any research project that generates a list of genes can take advantage of this visualization framework. Enrichment Map is implemented as a freely available and user friendly plug-in for the Cytoscape network visualization software (http://baderlab.org/Software/EnrichmentMap/).
Conflict of interest statement
Figures
References
-
- Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nature reviews Genetics. 2006;7:55–65. - PubMed
-
- Calarco JA, Saltzman AL, Ip JY, Blencowe BJ. Technologies for the global discovery and analysis of alternative splicing. Advances in experimental medicine and biology. 2007;623:64–84. - PubMed
-
- Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nature methods. 2007;4:787–797. - PubMed
-
- Quackenbush J. Computational analysis of microarray data. Nature reviews Genetics. 2001;2:418–427. - PubMed
-
- Nam D, Kim S-Y. Gene-set approach for expression pattern analysis. Briefings in bioinformatics. 2008;9:189–197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
