

Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding
- PMID: 21085610
- PMCID: PMC2978725
- DOI: 10.1371/journal.ppat.1001186
Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding
Abstract
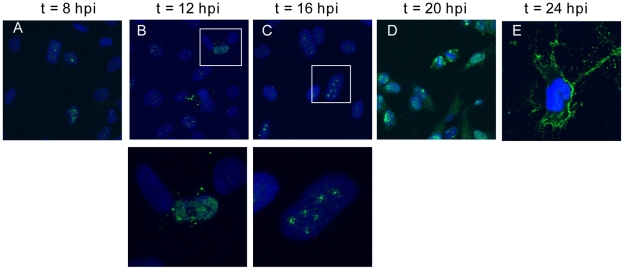
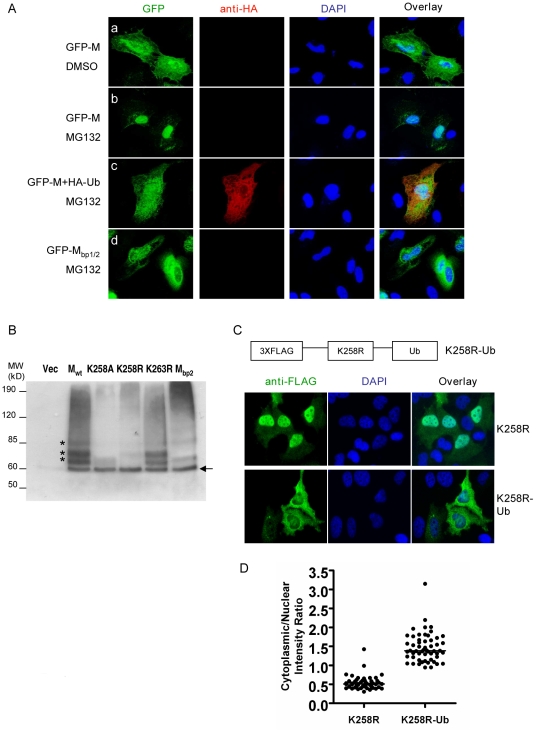
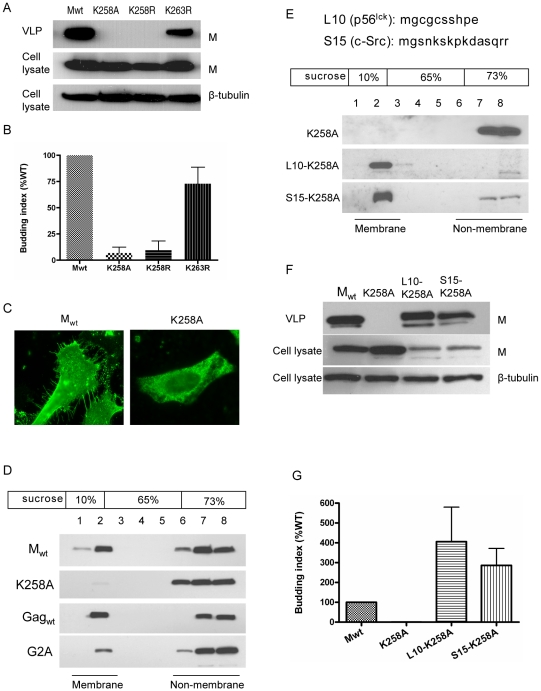
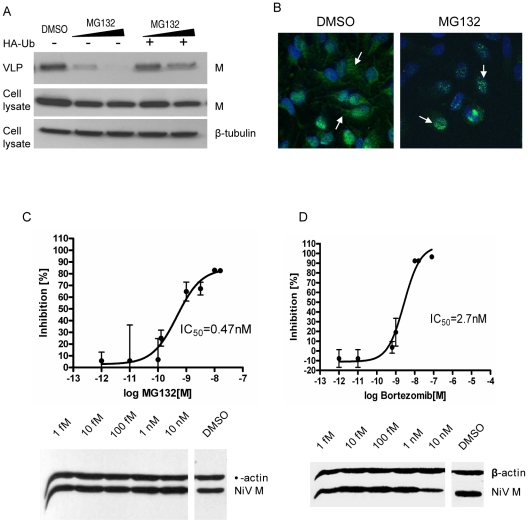
Paramyxoviruses are known to replicate in the cytoplasm and bud from the plasma membrane. Matrix is the major structural protein in paramyxoviruses that mediates viral assembly and budding. Curiously, the matrix proteins of a few paramyxoviruses have been found in the nucleus, although the biological function associated with this nuclear localization remains obscure. We report here that the nuclear-cytoplasmic trafficking of the Nipah virus matrix (NiV-M) protein and associated post-translational modification play a critical role in matrix-mediated virus budding. Nipah virus (NiV) is a highly pathogenic emerging paramyxovirus that causes fatal encephalitis in humans, and is classified as a Biosafety Level 4 (BSL4) pathogen. During live NiV infection, NiV-M was first detected in the nucleus at early stages of infection before subsequent localization to the cytoplasm and the plasma membrane. Mutations in the putative bipartite nuclear localization signal (NLS) and the leucine-rich nuclear export signal (NES) found in NiV-M impaired its nuclear-cytoplasmic trafficking and also abolished NiV-M budding. A highly conserved lysine residue in the NLS served dual functions: its positive charge was important for mediating nuclear import, and it was also a potential site for monoubiquitination which regulates nuclear export of the protein. Concordantly, overexpression of ubiquitin enhanced NiV-M budding whereas depletion of free ubiquitin in the cell (via proteasome inhibitors) resulted in nuclear retention of NiV-M and blocked viral budding. Live Nipah virus budding was exquisitely sensitive to proteasome inhibitors: bortezomib, an FDA-approved proteasome inhibitor for treating multiple myeloma, reduced viral titers with an IC(50) of 2.7 nM, which is 100-fold less than the peak plasma concentration that can be achieved in humans. This opens up the possibility of using an "off-the-shelf" therapeutic against acute NiV infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among Paramyxovirinae matrix proteins.PLoS Pathog. 2015 Mar 17;11(3):e1004739. doi: 10.1371/journal.ppat.1004739. eCollection 2015 Mar. PLoS Pathog. 2015. PMID: 25782006 Free PMC article.
-
The E3 ligase RAD18-mediated ubiquitination of henipavirus matrix protein promotes its nuclear-cytoplasmic trafficking and viral egress.Emerg Microbes Infect. 2025 Dec;14(1):2432344. doi: 10.1080/22221751.2024.2432344. Epub 2024 Dec 9. Emerg Microbes Infect. 2025. PMID: 39628457 Free PMC article.
-
Ubiquitination on Lysine 247 of Newcastle Disease Virus Matrix Protein Enhances Viral Replication and Virulence by Driving Nuclear-Cytoplasmic Trafficking.J Virol. 2022 Jan 26;96(2):e0162921. doi: 10.1128/JVI.01629-21. Epub 2021 Oct 27. J Virol. 2022. PMID: 34705566 Free PMC article.
-
Nipah virus matrix protein: expert hacker of cellular machines.FEBS Lett. 2016 Aug;590(15):2494-511. doi: 10.1002/1873-3468.12272. Epub 2016 Jul 12. FEBS Lett. 2016. PMID: 27350027 Free PMC article. Review.
-
Envelope-receptor interactions in Nipah virus pathobiology.Ann N Y Acad Sci. 2007 Apr;1102(1):51-65. doi: 10.1196/annals.1408.004. Ann N Y Acad Sci. 2007. PMID: 17470911 Free PMC article. Review.
Cited by
-
The ubiquitination of the influenza A virus PB1-F2 protein is crucial for its biological function.PLoS One. 2015 Apr 13;10(4):e0118477. doi: 10.1371/journal.pone.0118477. eCollection 2015. PLoS One. 2015. PMID: 25866881 Free PMC article.
-
Nuclear export signal and immunodominant CD8+ T cell epitope in influenza A virus matrix protein 1.J Virol. 2012 Sep;86(18):10258; author reply1259-60. doi: 10.1128/JVI.00894-12. J Virol. 2012. PMID: 22923812 Free PMC article. No abstract available.
-
Nipah Virus C Protein Recruits Tsg101 to Promote the Efficient Release of Virus in an ESCRT-Dependent Pathway.PLoS Pathog. 2016 May 20;12(5):e1005659. doi: 10.1371/journal.ppat.1005659. eCollection 2016 May. PLoS Pathog. 2016. PMID: 27203423 Free PMC article.
-
Importin α5 negatively regulates importin β1-mediated nuclear import of Newcastle disease virus matrix protein and viral replication and pathogenicity in chicken fibroblasts.Virulence. 2018 Dec 31;9(1):783-803. doi: 10.1080/21505594.2018.1449507. Virulence. 2018. PMID: 29532715 Free PMC article.
-
ANP32B is a nuclear target of henipavirus M proteins.PLoS One. 2014 May 13;9(5):e97233. doi: 10.1371/journal.pone.0097233. eCollection 2014. PLoS One. 2014. PMID: 24823948 Free PMC article.
References
-
- Field H, Young P, Yob JM, Mills J, Hall L, et al. The natural history of Hendra and Nipah viruses. Microbes Infect. 2001;3:307–314. - PubMed
-
- Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, et al. Nipah virus: a recently emergent deadly paramyxovirus. Science. 2000;288:1432–1435. - PubMed
-
- Weingartl HM, Berhane Y, Czub M. Animal models of henipavirus infection: a review. Vet J. 2009;181:211–220. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
