

High sensitivity TSS prediction: estimates of locations where TSS cannot occur
- PMID: 21085627
- PMCID: PMC2981523
- DOI: 10.1371/journal.pone.0013934
High sensitivity TSS prediction: estimates of locations where TSS cannot occur
Abstract
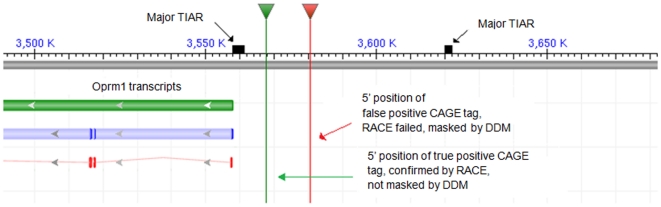
Background: Although transcription in mammalian genomes can initiate from various genomic positions (e.g., 3'UTR, coding exons, etc.), most locations on genomes are not prone to transcription initiation. It is of practical and theoretical interest to be able to estimate such collections of non-TSS locations (NTLs). The identification of large portions of NTLs can contribute to better focusing the search for TSS locations and thus contribute to promoter and gene finding. It can help in the assessment of 5' completeness of expressed sequences, contribute to more successful experimental designs, as well as more accurate gene annotation.
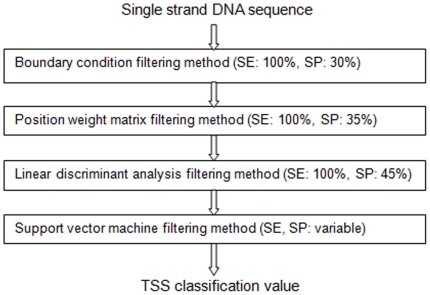
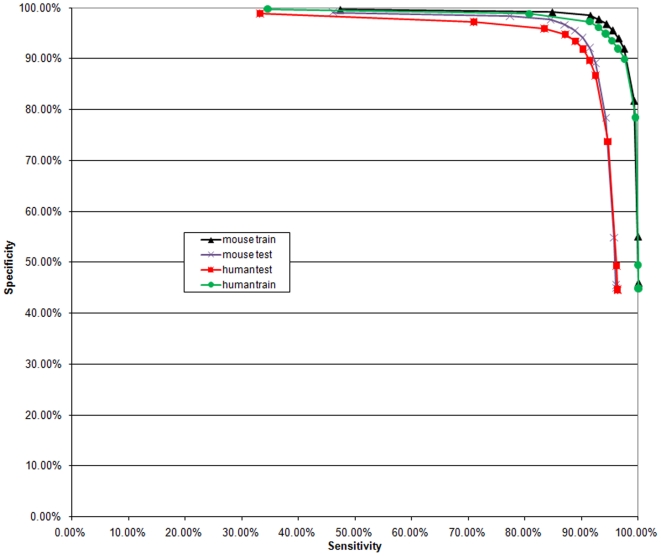
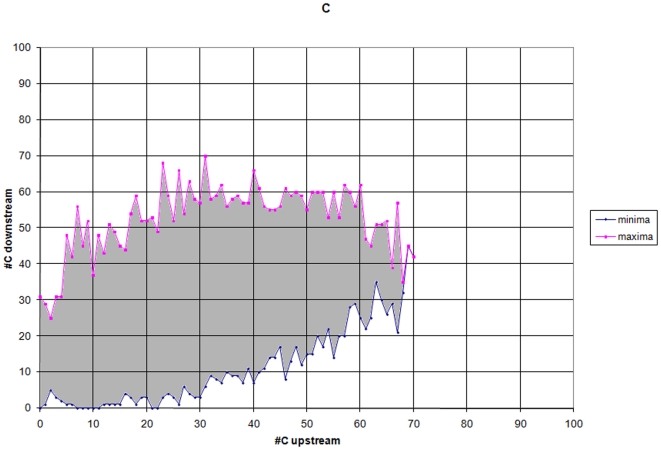
Methodology: Using comprehensive collections of Cap Analysis of Gene Expression (CAGE) and other transcript data from mouse and human genomes, we developed a methodology that allows us, by performing computational TSS prediction with very high sensitivity, to annotate, with a high accuracy in a strand specific manner, locations of mammalian genomes that are highly unlikely to harbor transcription start sites (TSSs). The properties of the immediate genomic neighborhood of 98,682 accurately determined mouse and 113,814 human TSSs are used to determine features that distinguish genomic transcription initiation locations from those that are not likely to initiate transcription. In our algorithm we utilize various constraining properties of features identified in the upstream and downstream regions around TSSs, as well as statistical analyses of these surrounding regions.
Conclusions: Our analysis of human chromosomes 4, 21 and 22 estimates ∼46%, ∼41% and ∼27% of these chromosomes, respectively, as being NTLs. This suggests that on average more than 40% of the human genome can be expected to be highly unlikely to initiate transcription. Our method represents the first one that utilizes high-sensitivity TSS prediction to identify, with high accuracy, large portions of mammalian genomes as NTLs. The server with our algorithm implemented is available at http://cbrc.kaust.edu.sa/ddm/.
Conflict of interest statement
Figures
Similar articles
-
Dragon gene start finder: an advanced system for finding approximate locations of the start of gene transcriptional units.Genome Res. 2003 Aug;13(8):1923-9. doi: 10.1101/gr.869803. Epub 2003 Jul 17. Genome Res. 2003. PMID: 12869582 Free PMC article.
-
TSSFinder-fast and accurate ab initio prediction of the core promoter in eukaryotic genomes.Brief Bioinform. 2021 Nov 5;22(6):bbab198. doi: 10.1093/bib/bbab198. Brief Bioinform. 2021. PMID: 34050351 Free PMC article.
-
Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis - a rich resource to identify new transcripts, proteins and to study gene regulation.BMC Genomics. 2016 Apr 23;17:302. doi: 10.1186/s12864-016-2602-9. BMC Genomics. 2016. PMID: 27107716 Free PMC article.
-
Computational annotation of miRNA transcription start sites.Brief Bioinform. 2021 Jan 18;22(1):380-392. doi: 10.1093/bib/bbz178. Brief Bioinform. 2021. PMID: 32003428 Free PMC article. Review.
-
Role of DNA sequence based structural features of promoters in transcription initiation and gene expression.Curr Opin Struct Biol. 2014 Apr;25:77-85. doi: 10.1016/j.sbi.2014.01.007. Epub 2014 Feb 4. Curr Opin Struct Biol. 2014. PMID: 24503515 Review.
Cited by
-
Alternative transcribed 3' isoform of long non-coding RNA Malat1 inhibits mouse retinal oxidative stress.iScience. 2022 Dec 5;26(1):105740. doi: 10.1016/j.isci.2022.105740. eCollection 2023 Jan 20. iScience. 2022. PMID: 36594014 Free PMC article.
-
Deep learning and support vector machines for transcription start site identification.PeerJ Comput Sci. 2023 Apr 17;9:e1340. doi: 10.7717/peerj-cs.1340. eCollection 2023. PeerJ Comput Sci. 2023. PMID: 37346545 Free PMC article.
-
Evolution of Brain Active Gene Promoters in Human Lineage Towards the Increased Plasticity of Gene Regulation.Mol Neurobiol. 2018 Mar;55(3):1871-1904. doi: 10.1007/s12035-017-0427-4. Epub 2017 Feb 24. Mol Neurobiol. 2018. PMID: 28233272
-
Critical assessment of computational tools for prokaryotic and eukaryotic promoter prediction.Brief Bioinform. 2022 Mar 10;23(2):bbab551. doi: 10.1093/bib/bbab551. Brief Bioinform. 2022. PMID: 35021193 Free PMC article.
-
Computational identification of eukaryotic promoters based on cascaded deep capsule neural networks.Brief Bioinform. 2021 Jul 20;22(4):bbaa299. doi: 10.1093/bib/bbaa299. Brief Bioinform. 2021. PMID: 33227813 Free PMC article.
References
-
- Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15776–81. doi: 10.1073/pnas.2136655100. - DOI - PMC - PubMed
-
- Ng P, Wei C, Sung W, Chiu KP, Lipovich L, et al. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nature methods. 2005;2:105–11. doi: 10.1038/nmeth733. - DOI - PubMed
-
- Wei C, Ng P, Chiu KP, Wong CH, Ang CC, et al. 5′ Long serial analysis of gene expression (LongSAGE) and 3′ LongSAGE for transcriptome characterization and genome annotation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11701–6. doi: 10.1073/pnas.0403514101. - DOI - PMC - PubMed
-
- Hashimoto S, Suzuki Y, Kasai Y, Morohoshi K, Yamada T, et al. 5′-end SAGE for the analysis of transcriptional start sites. Nature biotechnology. 2004;22:1146–9. doi: 10.1038/nbt998. - DOI - PubMed
-
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, et al. The transcriptional landscape of the mammalian genome. Science (New York, N.Y.) 2005;309:1559–63. doi: 10.1126/science.1112014. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
