

The role of oligomerization and cooperative regulation in protein function: the case of tryptophan synthase
- PMID: 21085641
- PMCID: PMC2978696
- DOI: 10.1371/journal.pcbi.1000994
The role of oligomerization and cooperative regulation in protein function: the case of tryptophan synthase
Abstract
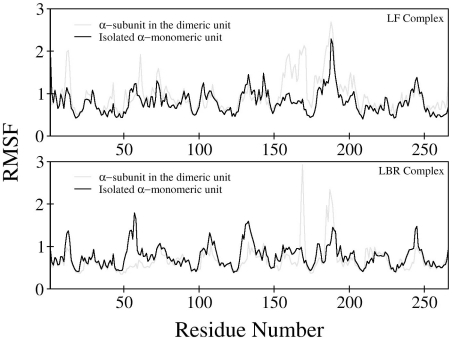
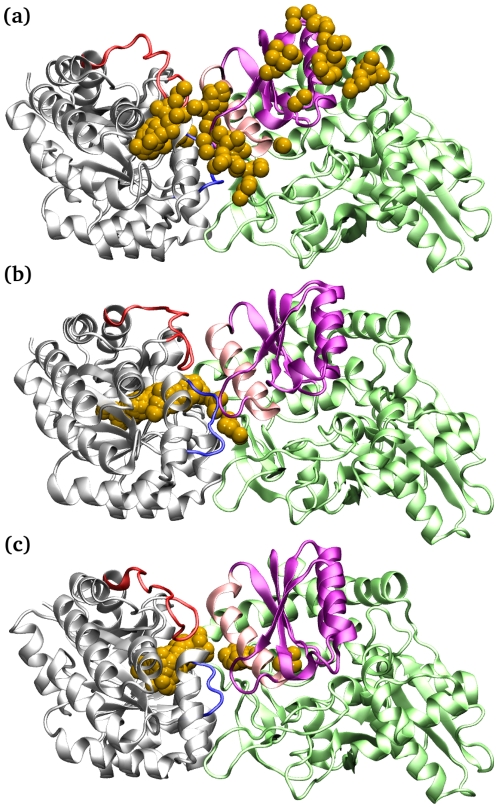
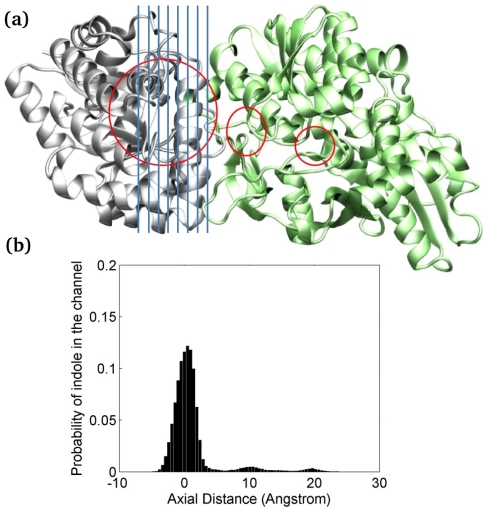
The oligomerization/co-localization of protein complexes and their cooperative regulation in protein function is a key feature in many biological systems. The synergistic regulation in different subunits often enhances the functional properties of the multi-enzyme complex. The present study used molecular dynamics and Brownian dynamics simulations to study the effects of allostery, oligomerization and intermediate channeling on enhancing the protein function of tryptophan synthase (TRPS). TRPS uses a set of α/β-dimeric units to catalyze the last two steps of L-tryptophan biosynthesis, and the rate is remarkably slower in the isolated monomers. Our work shows that without their binding partner, the isolated monomers are stable and more rigid. The substrates can form fairly stable interactions with the protein in both forms when the protein reaches the final ligand-bound conformations. Our simulations also revealed that the α/β-dimeric unit stabilizes the substrate-protein conformation in the ligand binding process, which lowers the conformation transition barrier and helps the protein conformations shift from an open/inactive form to a closed/active form. Brownian dynamics simulations with a coarse-grained model illustrate how protein conformations affect substrate channeling. The results highlight the complex roles of protein oligomerization and the fine balance between rigidity and dynamics in protein function.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- Woolf PJ, Linderman JJ. Self organization of membrane proteins via dimerization. Biophys Chem. 2003;104:217–227. - PubMed
-
- Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618–625. - PubMed
-
- Meng GY, Fronzes R, Chandran V, Remaut H, Waksman G. Protein oligomerization in the bacterial outer membrane. Mol Membr Biol. 2009;26:136–145. - PubMed
-
- Ali MH, Imperiali B. Protein oligomerization: How and why. Bioorgan Med Chem. 2005;13:5013–5020. - PubMed
-
- Brinda KV, Vishveshwara S. Oligomeric protein structure networks: insights into protein-protein interactions. BMC Bioinformatics. 2005;6 doi: 10.1186/1471-2105-6-296. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
